





Abstract
The Precision Medicine Initiative is a multicenter effort aiming at formulating personalized treatments leveraging on individual patient data (clinical, genome sequence and functional genomic data) together with the information in large knowledge bases (KBs) that integrate genome annotation, disease association studies, electronic health records and other data types. The biomedical literature provides a rich foundation for populating these KBs, reporting genetic and molecular interactions that provide the scaffold for the cellular regulatory systems and detailing the influence of genetic variants in these interactions. The goal of BioCreative VI Precision Medicine Track was to extract this particular type of information and was organized in two tasks: (i) document triage task, focused on identifying scientific literature containing experimentally verified protein–protein interactions (PPIs) affected by genetic mutations and (ii) relation extraction task, focused on extracting the affected interactions (protein pairs). To assist system developers and task participants, a large-scale corpus of PubMed documents was manually annotated for this task. Ten teams worldwide contributed 22 distinct text-mining models for the document triage task, and six teams worldwide contributed 14 different text-mining systems for the relation extraction task. When comparing the text-mining system predictions with human annotations, for the triage task, the best F-score was 69.06%, the best precision was 62.89%, the best recall was 98.0% and the best average precision was 72.5%. For the relation extraction task, when taking homologous genes into account, the best F-score was 37.73%, the best precision was 46.5% and the best recall was 54.1%. Submitted systems explored a wide range of methods, from traditional rule-based, statistical and machine learning systems to state-of-the-art deep learning methods. Given the level of participation and the individual team results we find the precision medicine track to be successful in engaging the text-mining research community. In the meantime, the track produced a manually annotated corpus of 5509 PubMed documents developed by BioGRID curators and relevant for precision medicine. The data set is freely available to the community, and the specific interactions have been integrated into the BioGRID data set. In addition, this challenge provided the first results of automatically identifying PubMed articles that describe PPI affected by mutations, as well as extracting the affected relations from those articles. Still, much progress is needed for computer-assisted precision medicine text mining to become mainstream. Future work should focus on addressing the remaining technical challenges and incorporating the practical benefits of text-mining tools into real-world precision medicine information-related curation.
Introduction and motivation
BioCreative challenges (1–8), historically, have aimed to bring forth community tasks that result in the development of text-mining systems that can be of practical use to database curators and the users of textual data in the field of biology. The choice of tasks has covered identification of biologically relevant entities such as genes, proteins, species, diseases and chemicals, as well as their interactions in biomedical literature. These tasks have researched important factors about usability and understanding curation workflows (8–12), have focused on building text-mining systems that address users’ requirements (8–10) as well as foster standard developments for issues of use, reuse and integration (7, 13, 14). In addition, keeping with the current needs, community challenges in biomedical natural language processing such as BioNLP and BioASQ (15–18) have addressed development of information extraction systems for relevant and emerging research areas. Finally, all these tasks have also provided and produced quality data sets for training and testing of automated systems that contained abstracts of biomedical scientific publications, as well as full text (19–25).
Precision medicine is an emerging approach for disease treatment and prevention that takes into account variability in genes, environment and lifestyle for each person. This emerging research area requires interdisciplinary collaboration between different fields such as medical practitioners, medical informaticians, biomedical researchers and data analytics. Precision medicine has demonstrated great promise in the field of cancer medicine, which led to the near-term focus of the US Precision Medicine Initiative being cancer diagnosis and treatment (26). To efficiently translate this new approach into clinical practice it is required to foster the de novo development and access to knowledge bases (KBs) storing and organizing the potential effect of genetic variations on molecular phenotypes.
One area of great relevance is the study of cellular networks, which underlie the structure and the function of the cell (27). Understanding how genetic variation can affect interaction stability between gene pairs and how this variation can influence the response of cellular pathways at an individual level is crucial for the goals of precision medicine. With this in mind, we organized a novel challenge in BioCreative VI aiming at creating automated systems capable of extracting such information from the scientific literature for supporting precision medicine.
Text mining and natural language processing have an intuitive place in the framework for the implementation of precision medicine (28, 29). Much of the required information, about genes/proteins, mutations, diseases and their interactions, can be found in the unstructured text of scientific articles indexed in PubMed (28, 30–35). Specialized curation databases, such as IntAct (1, 36) and BioGRID (37), have been collecting and cataloging knowledge focused on particular areas of biology since 2004, so that they may enable insights into conserved networks and pathways that are relevant to human health. Expanding their curation efforts into capturing specific sequence-variant-depended molecular interactions may open up new possibilities and enable insights that pertain to precision medicine. To date, no tool is available to facilitate this kind of specific retrieval. Therefore, the goal of the precision medicine track in BioCreative VI was to foster the development of text-mining algorithms that specialize in scanning the published biomedical literature to extract the reported discoveries of protein interactions changing in nature due to the presence of genomic variations or artificial mutations.
Information retrieval related to precision medicine has also been explored by the 2017 TREC Precision Medicine Track (http://www.trec-cds.org/2017.html). Differently from our challenge, the TREC challenge focused on ranking PubMed articles and clinical trials for synthetic case patients described as a set of relevant facts such as disease type, genetic variant and basic demographic information. Participants in the TREC challenge were asked to rank PubMed articles addressing relevant treatments for the given patient and clinical trials for which the patient could be eligible. The goal was to foster algorithms specialized in retrieval of existing treatments from the current scientific literature as well as to identify the potential for experimental treatments.
Prioritizing articles for manual literature curation is crucial in the climate of exponential growth of published articles. Assessment of retrieval algorithms for annotation databases has been studied in detail in the context of TREC Genomics tracks and several BioCreative challenges. The resulting text-mining tools and other tools inspired by these challenges and/or trained on the data sets produced via these challenges, such as BioQRator (38, 39) and PubTator (40), are routinely used by curators to simplify the identification of relevant articles for curation from a range of journals that publish protein interaction studies.
In order for computers to aid manual curation, annotated data sets need to be available for computer algorithm development and evaluation. To this end, we identified a challenge track of retrieving and extracting precision medicine-relevant information from PubMed articles. Specifically, we focused on (i) the identification of scientific literature pertinent to protein interaction and mutation and (ii) extraction of proteins whose interactions are affected in the presence of a sequence variation. To address this, we proposed two challenge tasks:
Document triage task: identification and ranking of relevant PubMed citations describing protein–protein interactions (PPIs) affected by mutations. The goal of this task is to foster development of highly accurate information retrieval algorithms that prioritize these articles for database curation. The algorithms are expected to scan the title and abstract of a PubMed document and predict its relevance for curation for precision medicine purposes. A typical article is shown in Figure 1.
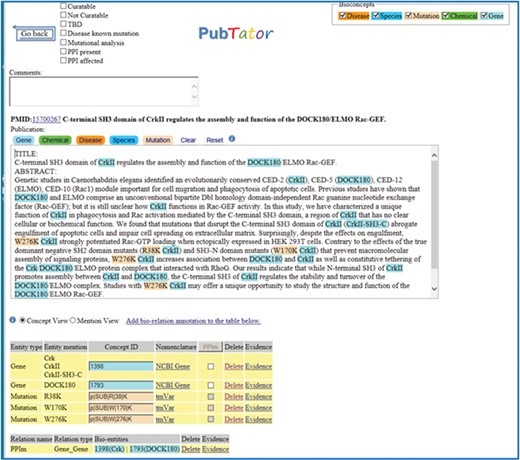
A positive example from the Biocreative VI Precision Medicine Track corpus.
PPI extraction has been the focus of many previous challenges in biomedical text mining. However, this new task prioritized the selection of articles reporting the effect of sequence variation (mutation/deletion) on experimentally verified PPI interactions. Oftentimes, the article abstract does not name the mutation or permit the unambiguous assignment of database identifiers to the interacting genes. However, if the article abstract describes evidence suggesting to the curator that the specific information is in the full text, that document is still considered relevant for triage. For this task, participants were given as input the articles’ title and abstract, as well as the recognized genes via available text-mining systems. For system output, organizers asked participants to predict a label (relevant/not relevant) for each article in the test set and submit confidence scores for their predictions [in the range (0,1)] to facilitate the ranking of results. Manual annotations were used as gold standard for evaluating team submissions. Each team was allowed to submit three runs.
Relation extraction task: extraction of experimentally verified PPI pairs affected by the presence of a genetic mutation.
The goal of this task is to foster development of highly accurate information extraction algorithms that can facilitate curation of precision medicine related information. The algorithms are expected to scan the title and abstract of a PubMed article and determine the interacting pair of proteins whose interaction is affected by a genetic mutation, as experimentally verified in the article.
This task is a step toward the ultimate goal of using computers for assisting human curation. As in the previous task, participants were provided with the articles’ title and abstract, as well as the recognized genes via available text-mining systems. For system output, participants were asked to return interacting protein pairs affected by mutations. Each pair is described by their Entrez Gene ID. Manually curated interacting pairs were used as the gold standard for evaluating team predictions. Each team was allowed to submit three runs.
Generally speaking, the first task is an information retrieval task, while the second task can be categorized as an information extraction task.
Methods and data
To achieve this, we needed to create a large-scale data set to be used for article triage and a smaller data set to be utilized for the evaluation of the relation extraction. Here we describe how we developed the track and give some details on the evaluation procedure and corpus annotation.
Statistics of the precision medicine track data set
| Data set | Articles | Positive | Negative | Articles with relations | Number of relations |
|---|---|---|---|---|---|
| Training | 4082 | 1729 | 2353 | 597 | 752 |
| Testing | 1427 | 704 | 723 | 635 | 869 |
Precision Medicine Track corpus development and annotation
The first step in developing an automated text-mining system that extracts specialized information is the curation of a manually annotated corpus that could be used for the training, tuning and development of such algorithms. Our research on creating and developing our training corpus (19) showed that biomedical literature is ripe with precision medicine-relevant information.
Five professional BioGRID curators contributed to the development of the Precision Medicine Track corpus (corpus available from https://biocreative.bioinformatics.udel.edu/tasks/biocreative-vi/track-4/). The corpus is composed of two collections: the training data set and the testing data set, as shown in Table 1 and was used for training and evaluation of both triage and relation extraction tasks. The training set (4082 PubMed abstracts) creation consisted of manually reviewing PubMed abstracts from the following two different sources: expert curated databases (https://www.ebi.ac.uk/intact/) and state-of-the-art text-mining tools [PubMed articles were scored and ranked using PIE `the search’ (41), and the mutation mentions were identified via tmVar (35, 42, 43)]. As previously described in (19) each of these PubMed documents was first manually labeled for relevance for the triage task, and next, for the relation extraction task, the subset of PubMed documents that had been previously curated by IntAct/Mint for PPI relations was annotated with those interacting protein pairs if the interactions were affected by mutations, and the interaction was named in the abstract. A relation annotation consisted of a pair of interacting proteins Entrez Gene (https://www.ncbi.nlm.nih.gov/gene/) IDs. A typical article and its annotations in the Precision Medicine Track data set are shown in Figure 1.
The testing data set consisted of 1427 articles that contain the same ratio of positive to negative articles as the training data set and similar overlap of organisms, as discussed in the Precision Medicine Track corpus paper (44). These articles were not previously annotated in any public curation database. Therefore, for optimal annotation, the documents in the testing data set were distributed to five BioGRID curators such that each document was annotated by at least two curators. The PubTator tool was modified to assist the curators for this purpose. To annotate the testing data set, each curator was asked, in addition to their routine curation of PPI information for BioGRID, to visit the PubTator site where the articles were loaded for their manual annotation. For each article, curators marked ‘relevant’ if it described PPIs affected by mutations, ‘not relevant’ otherwise, and if ‘relevant’, they identified the interacting pair of proteins with their Entrez Gene ID, if the interaction was mentioned in the title and/or abstract.
For obtaining high-quality and consistent annotations across curators, detailed annotation guidelines were developed. In addition, all curators were asked to annotate a small test set, before being paired up for sets of 100 articles. Curator pairs were assigned randomly and every curator was paired at least twice with every other curator. After annotating a set of articles, curators met regularly to discuss and resolve their differences so that the final corpus is produced with complete consensus from all curators (44).
Track development and evaluation measures
The general evaluation setting for the Precision Medicine Track was to provide training data to participating teams, as well as the evaluation software that is available at https://github.com/ncbi-nlp/BC6PM. Data and annotations were produced in BioC (28) format (XML/JSON). The evaluation scripts also served as a self-check to ensure that the data output was in the correct format for evaluation. During this phase, teams had 3 months to implement their systems and improve them using the provided data. In the case that difficulties or unclear aspects were encountered, they could contact the organizers directly or use the group email list where information about the task was posted periodically. Organizers also set up a PubTator (https://www.ncbi.nlm.nih.gov/CBBresearch/Lu/Demo/PubTator/) (29) view, so that track participants could visualize the training data annotations. All participant teams were allowed to use provided text-mining systems (such as PubTator) and others to obtain automatically predicted bio-entities in the training data, if desired.
Evaluation measures for both tasks were standard evaluation procedures precision, recall, F-score and average precision. The evaluation software takes as input a set of two files: a system output file of a participating run (each participating team could submit up to three system outputs or runs) and a reference file consisting of manual annotations. For the document triage task, participants were asked to predict a label (relevant/not relevant) for each article in the test set and to submit confidence scores for their predictions [in the range (0,1)], which facilitated the ranking of results. The evaluation procedure measured each system’s ability to provide the best possible ranked list of relevant abstracts sorted from most relevant to the most irrelevant article.
For the relation extraction task, participants were asked to submit pairs of gene identifiers to denote PPI relations that are affected by mutations. For this task, organizers employed a two-level evaluation and calculated precision, recall and F-measure at the micro and macro level in two ways:
Exact match: a strict measure that requires that predicted relations exactly match the human annotations. In this scenario, all system-predicted relations (pairs of interacting proteins) were checked against the manually annotated ones for correctness. In our annotation format, a PPI relation is not defined as directional, therefore the order of interacting genes is not considered when checking for a match, i.e. relation(A, B) is equivalent to relation(B, A).
HomoloGene (https://www.ncbi.nlm.nih.gov/homologene) match: a more relaxed measure where a prediction is considered correct, as long as the system-predicted gene identifiers were homologous to the ones in the gold standard according to the HomoloGene database. In this scenario, all gene identifiers in the predicted relations and manually annotated data were mapped to common identifiers representing common HomoloGene classes, then all predicted relations were checked for correctness. If the predicted Gene IDs and the annotated Gene IDs in a relation were homologous genes, they were counted as a match. This evaluation was included to allow for the difficulty of mapping the correct Gene Identifier using only the abstract data. Often, authors include the specific details such as the organism/species in the full text.
Benchmarking systems
For comparison purposes, we developed a baseline method for both triage and relation extraction tasks. For the triage task, we designed a baseline Support Vector Machine (SVM) classifier using unigram and bigram features from titles and abstracts of the training corpus (19). For the relation extraction task, we implemented a simple co-occurrence baseline method. The Gene entities in the PubMed articles were automatically recognized using GNormPlus, SR4GN and tmVar (35, 42, 43, 45, 46), and a relation was predicted if two gene entities were found in the same sentence, regardless of whether a sequence variant had been predicted for that article or not.
Results
Precision Medicine Track corpus
All track participant teams were provided with the training data set for training their algorithms and with the PubMed articles in the testing data set to return their predictions. Our evaluation for the two tasks was to assess the teams’ ability to return relevant articles and the relevant PPI interactions when an article was describing protein interactions affected by specific mutations. Participants were not required to name the sequence variant, as, during the corpus annotation phase it was discovered that such information was not often specified in the PubMed article abstract. It was also observed that, in some articles, the interacting proteins were not directly specified in the title or abstract. Hence, Table 1 shows the overall statistics for the Precision Medicine Track corpus, including the number of relevant articles, number of relevant relations, as well as number of relevant articles with relevant relations. For example, relations of interacting proteins affected by the presence of a mutation were recorded as interacting pairs in 635 of the 704 articles marked relevant in the testing data set. Certain articles, while relevant for curation, need full-text perusal for the precise identification of the affected PPI.
Team participation results and discussion
Overall, 11 teams participated in the Precision Medicine Track, 10 teams in the document triage task and 6 teams in the relation extraction task. Since each team could submit up to three runs (i.e. three different versions of their system or contribute three different methods) for each task, a total of 36 runs were submitted. The participants were from Australia, China, Turkey, Greece, Portugal and the United States, as shown in Table 2.
Participating teams and their number of submissions
| Team number | Institution | Country | Triage task | Relation task |
|---|---|---|---|---|
| 374 | University of Aveiro | Portugal | 3 | - |
| 375 | University of Melbourne | Australia | 3 | 3 |
| 379 | Mayo Clinic | USA | 1 | 2 |
| 391 | Dalian University of Technology | China | - | 3 |
| 405 (Team withdrew) | - | - | 1 | 2 |
| 414 | Boğaziçi University | Turkey | 3 | - |
| 418 | National Technical University of Athens | Greece | 3 | - |
| 419 | Taipei Medical University | Taiwan | 3 | - |
| 420 | University of Kentucky | USA | 1 | 3 |
| 421 | Dalian University of Technology | China | 3 | - |
| 433 | Florida State University | USA | 1 | 1 |
For the triage task, we received results of 22 systems (shown in Table 3), 16 of which outperformed our baseline in F-score, 13 on average precision, 2 on precision and 17 on recall. The best F-score is 69.1%, the best average precision is 72.5%, the best precision is 62.9% and the best recall is 98.1%. The average F-score, average precision, precision and recall were 64.1%, 63.5%, 56.4% and 75.8%, respectively.
Document triage task results for all submissions
| Team number | Submission | Avg prec | Precision | Recall | F1 | Data format |
|---|---|---|---|---|---|---|
| 374 | Run 1 | 0.6616 | 0.5864 | 0.8338 | 0.6886 | JSON |
| Run 2 | 0.6677 | 0.5700 | 0.8736 | 0.6898 | JSON | |
| Run 3 | 0.6929 | 0.6070 | 0.7898 | 0.6864 | JSON | |
| 375 | Run 1 | 0.6822 | 0.5783 | 0.7713 | 0.6610 | JSON |
| Run 2 | 0.6722 | 0.5936 | 0.7116 | 0.6473 | JSON | |
| Run 3 | 0.6744 | 0.5361 | 0.8849 | 0.6677 | JSON | |
| 379 | Run 1 | 0.4904 | 0.4649 | 0.3480 | 0.3981 | XML |
| 405 | Run 1 | 0.5871 | 0.5484 | 0.5710 | 0.5595 | JSON |
| 414 | Run 1 | 0.4847 | 0.4734 | 0.5824 | 0.5223 | XML |
| Run 2 | 0.5057 | 0.4927 | 0.7202 | 0.5851 | XML | |
| Run 3 | 0.5077 | 0.5022 | 0.9801 | 0.6641 | XML | |
| 418 | Run 1 | 0.6959 | 0.6136 | 0.7670 | 0.6818 | XML |
| Run 2 | 0.7068 | 0.5944 | 0.8139 | 0.6871 | XML | |
| Run 3 | 0.7158 | 0.6289 | 0.7656 | 0.6906 | XML | |
| 419 | Run 1 | 0.5797 | 0.5713 | 0.8253 | 0.6752 | XML |
| Run 2 | 0.5986 | 0.5865 | 0.6065 | 0.5964 | XML | |
| Run 3 | 0.6334 | 0.5992 | 0.6222 | 0.6105 | XML | |
| 420 | Run 1 | 0.6439 | 0.5438 | 0.8736 | 0.6703 | JSON |
| 421 | Run 1 | 0.6678 | 0.5850 | 0.8111 | 0.6798 | XML |
| Run 2 | 0.7253 | 0.6073 | 0.7997 | 0.6904 | XML | |
| Run 3 | 0.7084 | 0.5857 | 0.8352 | 0.6885 | XML | |
| 433 | Run 1 | 0.6632 | 0.5413 | 0.8835 | 0.6713 | JSON |
| BASELINE | - | 0.6515 | 0.6122 | 0.6435 | 0.6274 | - |
For the relations task, we received results from 14 systems, 8 of which outperformed the baseline based on the F-score, 10 on precision and 1 on recall, when evaluated on Exact Match (see Table 4).
Relation extraction task exact match results for all submissions
| System | Submission | Precision | Recall | F1 | Data format |
|---|---|---|---|---|---|
| 375 | Run 1 | 0.3506 | 0.3349 | 0.3426 | XML |
| Run 2 | 0.3506 | 0.3349 | 0.3426 | XML | |
| Run 3 | 0.4000 | 0.3084 | 0.3483 | XML | |
| 379 | Run 1 | 0.2602 | 0.0736 | 0.1148 | XML |
| Run 2 | 0.1015 | 0.5121 | 0.1694 | XML | |
| 391 | Run 1 | 0.2253 | 0.1887 | 0.2054 | XML |
| Run 2 | 0.2222 | 0.1772 | 0.1972 | XML | |
| Run 3 | 0.2306 | 0.1300 | 0.1663 | XML | |
| 405 | Run 1 | 0.0590 | 0.0196 | 0.0294 | JSON |
| Run 2 | 0.0692 | 0.0253 | 0.0371 | JSON | |
| 420 | Run 1 | 0.3555 | 0.2336 | 0.2819 | JSON |
| Run 2 | 0.3739 | 0.2509 | 0.3003 | JSON | |
| Run 3 | 0.3494 | 0.2417 | 0.2857 | JSON | |
| 433 | Run 1 | 0.0580 | 0.2014 | 0.0900 | JSON |
| BASELINE | - | 0.1091 | 0.4741 | 0.1774 |
The HomoloGene evaluation showed a slightly different result: 6 systems outperformed the baseline on F-score, 10 on precision and 1 on recall. The average F-score, precision and recall for the HomoloGene evaluation were 23.8%, 28.1% and 24.5%, respectively. The best F-score, precision and recall were 37.7%, 46.5% and 54.1%, respectively. These results are shown in Table 5.
Relation extraction task HomoloGene results for all submissions
| System | Submission | Precision | Recall | F1 | Data format |
|---|---|---|---|---|---|
| 375 | Run 1 | 0.3807 | 0.3573 | 0.3686 | XML |
| Run 2 | 0.3807 | 0.3573 | 0.3686 | XML | |
| Run 3 | 0.4318 | 0.3341 | 0.3767 | XML | |
| 379 | Run 1 | 0.3102 | 0.0777 | 0.1243 | XML |
| Run 2 | 0.1160 | 0.5406 | 0.1910 | XML | |
| 391 | Run 1 | 0.2348 | 0.1972 | 0.2144 | XML |
| Run 2 | 0.2337 | 0.1868 | 0.2076 | XML | |
| Run 3 | 0.2398 | 0.1357 | 0.1733 | XML | |
| 405 | Run 1 | 0.0804 | 0.0267 | 0.0401 | JSON |
| Run 2 | 0.1044 | 0.0383 | 0.0560 | JSON | |
| 420 | Run 1 | 0.4417 | 0.2900 | 0.3501 | JSON |
| Run 2 | 0.4653 | 0.3109 | 0.3727 | JSON | |
| Run 3 | 0.4379 | 0.3028 | 0.3580 | JSON | |
| 433 | Run 1 | 0.0801 | 0.2749 | 0.1241 | JSON |
| BASELINE | - | 0.1468 | 0.5197 | 0.2290 |
The BioCreative VI Precision Medicine Track consisted of an information retrieval task and an information extraction task. The wide participation likewise resulted in a wide range of contributed algorithms such as various versions of deep learning methods: Convolutional Neural Networks (CNNs), Long Short-Term Memory (LSTM) neural networks and Hierarchical Attention Neural Networks, various statistical methods based on frequency of occurrence of specific terms or co-occurrence of entities, SVM models, Gradient Boosted Trees, etc. Teams also used additional resources to enrich training data such as PubMed article metadata (i.e. MeSH terms), previous BioCreative challenges PPI data sets, word embeddings trained on the whole MEDLINE, as well as to help their computation by utilizing in-house developed lists of terms to describe interactions and mutations, the Interaction Network Ontology terms, Genia Tagger, MEDLINE metadata and specialized PubMed search to extract links between PubMed articles and GENE database.
Useful model additions reported by participating teams were the use of the portion of the BioCreative III PPI data set labeled negative to enrich the current task training data set (University of Aveiro), Interaction Network Ontology terms (University of Bogaziçi), in-house developed special term lists (University of Melbourne), the use of MeSH terms (Taipei Medical University) to enrich the feature set, the use of five individual neural network models combined with majority voting, weighted voting and logistic regression (Dalian University) and the use of separate encoder architectures [bidirectional Recurrent Neural Networks(RNN)] for title and abstract (National Technical University of Athens), etc. to develop a more accurate model for better information retrieval.
The teams participating in the document triage task used the state-of-the-art methods in machine learning, focused on deep learning architecture (with few exceptions), and they achieved an average F-score of 64.08% and median F-score of 67.03%. These results are an improvement when compared with previous BioCreative tasks focused on retrieving articles for PPI, such as BioCreative III PPI Article Classification task, where the best reported F-score was 61.42% (47).
When compared with previous tasks in extracting relations from biomedical literature the results are as follows: BioCreative II PPI extraction with entity mapping to SwissProt IDs reported 39% as their highest achieved precision (47), and BioCreative V Chemical Disease Relation task (48) reported 55.67% as their highest precision in recognizing a chemical-induced disease relationship. The highest precision for the relation extraction task in our challenge was 40% for exact match and 46.53% for homology match.
The teams participating in relation extraction task reported lower results due to the difficulty of the task. Participating teams reported that there was a significant need for more accurate gene recognition tools, as standard bioNLP NER tools were not sufficient to identify all useful entities. To help with this, the team from University of Melbourne supplemented their system with manually defined term lists; the team from Dalian University developed in-house entity-relationship triplets extracted from public PPI databases; and the team from University of Kentucky used BioCreative II Gene Normalization lexicon, queried Entrez Gene database for each annotated gene mention in the training data and cross-referenced the result with a PMID-based query, verifying via Medline metadata.
To determine the difficulty of the relations tasks, we examined how many teams correctly identified each of the gold standard PPI relations in the test set. As shown in Table 6, 28.9% of the manually curated relations in the test set were not found by any of the teams. This means that only 71.11% of all relations could be extracted by at least one team. For those pairs, most (98%) were not present in the training set, and of the genes comprising those pairs, most (81.5%) were not present in the training set. In order to understand better, we randomly selected a sample of 10 PubMed documents with missed relations for manual inspection. For each of these documents, we looked at the relations that were marked by curators, and their HomoloGene counterparts, and the relations that were predicted by the participating teams for these documents, and their HomoloGene counterparts. This review identified two main reasons explaining why teams had difficulties extracting these relations: (i) one or both genes involved in the relation identified by curators were not found in the list of genes returned by the systems (even when correcting for homologous genes) and (ii) the evidence text describing the relation was not contained in one sentence and however was distributed throughout the abstract.
Overview of how many submissions correctly identified the protein interactions affected by mutations in the test set
| Relations in test set | ||
|---|---|---|
| Number of systems | Number | % |
| 0 | 249 | 28.89 |
| 1 | 81 | 9.40 |
| 2 | 46 | 5.34 |
| 3 | 115 | 13.34 |
| 4 | 96 | 11.14 |
| 5 | 55 | 6.38 |
| 6 | 39 | 4.52 |
| 7 | 56 | 6.50 |
| 8 | 59 | 6.84 |
| 9 | 18 | 2.09 |
| 10 | 22 | 2.55 |
| 11 | 18 | 2.09 |
| 12 | 3 | 0.35 |
| 13 | 4 | 0.46 |
| Sum | 869 | 100.00 |
In order to illustrate some of the different ways that this type of relation is expressed in PubMed literature, we list some examples from our corpus documents in Table 7. For each example we show the article identifier (PMID), the curated relation as a pair of Entrez Gene identifiers, the number of submissions that were able to extract that particular relation, as well as a text excerpt from the PubMed abstract that describes the relation. The text excerpt contains highlights of the gene mentions and, to facilitate the mapping with the curated values in Column 2, the corresponding gene identifiers are given in parenthesis. We have prepared a more detailed compilation of such examples, listing the complete abstract for each of the documents, in a Supplementary data.
Examples of relations in the test set. For each example we give the article identifier (PMID), the relation as extracted by curators specified as two Entrez Gene IDs, the number of systems that extracted that particular relation and a text excerpt from the corresponding abstract that describes the relation. The gene mentions are highlighted in the text excerpt, and the Entrez Gene IDs are given in parenthesis. The relations that have not been detected by systems are typically described in several sentences, describe the absence of an interaction with another protein or contain a self-interaction
| PMID | Relation | Number of systems | Text |
|---|---|---|---|
| 15700267 | 1398, 1793 | 13 | Contrary to the effects of the true dominant negative SH2 domain mutants (R38K CrkII) and SH3-N domain mutants (W170K CrkII) that prevent macromolecular assembly of signaling proteins, W276K CrkII increases association between DOCK180 (1793) and CrkII (1398) as well as constitutive tethering of the Crk/DOCK180/ELMO protein complex that interacted with RhoG. |
| 16969499 | 672, 7157 | 13 | Co-immunoprecipitation assays of Escherichia coli-expressed wild-type and mutated BRCTs challenged with a HeLa cell extract revealed, for the S1841 N variant a significant reduction in the binding activity to p53, while the W1837R mutant showed an inverse effect. Furthermore, a clonogenic soft agar growth assay performed on HeLa cells stably transfected with either wild-type or mutant BRCA1 showed a marked decrease of the growth in wild-type BRCA1-overexpressing cells and in BRCA1S1841N-transfected cells, while no significant changes were detected in the BRCA1W1837R-transfected cells. These results demonstrate that distinct single nucleotide changes in the BRCT domain of BRCA1 (672) affect binding of this protein to the tumor suppressor p53 (7157). |
| 11463845 | 1026, 207 | 5 | Here we demonstrate that Akt (207) phosphorylates the cell cycle inhibitory protein p21(Cip1) (1026) at Thr 145 in vitro and in intact cells as shown by in vitro kinase assays, site-directed mutagenesis and phospho-peptide analysis. |
| 9234717 | 12402, 18595 | 4 | In vitro, Cbl-N (12402) directly bound to PDGFR alpha (18595) derived from PDGF-AA-stimulated cells but not to that from unstimulated cells, and this binding was abrogated by a point mutation (G306E) corresponding to a loss-of-function mutation in SLI-1. |
| 16144832 | 300772, 60590 | 0 | Pias1(300772) binding to mGluR8-C60590 required a region N-terminal to a consensus sumoylation motif and was not affected by arginine substitution of the conserved lysine 882 within this motif. |
| 8623535 | 1489075, 1489080 | 0 | The E2 binding activity of E1 deletion and point mutant proteins were assayed using glutathione S-transferase E1 fusion proteins and in vitro translated proteins. At 4, the C-terminal portion of E1 (1489075) including amino acids 312–644 was sufficient for E2 (1489080) binding. Introduction of C-terminal deletions or a point mutation at position 586 (Pro → Glu) resulted in the loss of the E2 binding activity. |
| 14985338 | 6804, 9751 | 0 | cAMP-dependent protein kinase (PKA) can modulate synaptic transmission by acting directly on the neurotransmitter secretory machinery. Here we identify one possible target, syntaphilin, which was identified as a molecular clamp that controls free syntaxin-1 and dynamin-1 availability and thereby regulates synaptic vesicle exocytosis and endocytosis. Deletion mutation and site-directed mutagenesis experiments pinpoint dominant PKA phosphorylation sites to serines 43 and 56. PKA phosphorylation of syntaphilin significantly decreases its binding to syntaxin-1A (6804) in vitro. A syntaphilin (9751) mutation of serine 43 to aspartic acid (S43D) shows similar effects on binding. |
| 15769741 | 285, 285 | 0 | In addition, improper creation of a new cysteine in Ang2 (285) (Ang2S263C) dramatically induced Ang2 aggregation without activating Tie2. |
| 9099695 | 495516, 495516 | 0 | These mutants confirmed that Ser-190 is a major autophosphorylation site of Pim-1 (495516). |
| 9786907 | 1030, 1030 | 0 | Analytical centrifugation allowed to determine that p15 (1030) assembles as a rod-shaped tetramer. Oxidative cross-linking of N-terminal cysteines of the peptide generated specific covalent oligomers, indicating that the N terminus of p15 is a coiled coil that assembles as a parallel tetramer. Mutation of Lys22 into Asp destabilized the tetramer and put forward the presence of a salt bridge between Lys22 and Asp24 in a model building of the stalk. |
Individual system descriptions
All participating teams were requested to provide a short technical summary on the strategy used for participation in the Precision Medicine Track, which is listed below. Team summaries are ordered based on a team identifier.
Team 374: University of Aveiro
In the Precision Medicine document triage task, we employed a deep learning approach with combinations of convolutional and LSTM networks. We used pre-calculated word embeddings, trained on the complete MEDLINE database, corresponding to 15 million abstracts in English language. We used the word2vec implementation in the Gensim framework (59) to generate six models with vector sizes of 100 and 300 features and using windows of 5, 20 and 50. The models contain around 775 000 distinct words, and we used the model with 300 features and a window size of 50.
For the official participation in the task, we implemented two network architectures. The first was composed of the embedding layer with fixed weights, followed by three convolutional layers, each using 128 filters with a kernel size of three and the Rectified Linear Unit activation function. Average pooling over windows of size three was applied to the output of the third convolutional layers, and this was connected to a bidirectional LSTM layer with 128 units. Finally, a densely connected layer with a sigmoid activation function is used for classification. The second network was deeper, with three convolutional layers as in the first network but with the number of filters set to 64, followed by a bidirectional LSTM layer and two unidirectional LSTMs. All three LSTMs were composed of 128 units.
We also explored the use of the BioCreative III PPI-ACT corpus as additional data through a self-learning approach. This corpus consists of 12 280 Medline abstracts, 2732 of which were annotated as relevant for PPI information; however, these articles have not been annotated considering the impact of genetic mutations as expected for the current task. During our tests, including the negative documents produced small improvements in the results, while including positive documents (as per the BC-III guidelines) decreased the classifier performance.
Following the workshop, we implemented a shallower architecture, composed of a single convolutional layer, average pooling and a single LSTM layer, followed by an attention layer (49) and achieved similar results as obtained in the official evaluation. We also applied grid search in an attempt to optimize the hyper-parameters of the network, namely number of filters and percentage of dropout, but could not improve the results.
Team 375: University of Melbourne
The University of Melbourne READ-Biomed team participated in the document triage and relation extraction tasks of the Precision Medicine track of BioCreative VI. For the document triage task, we constructed term lists consisting of terms that are used to describe interactions, mutations and expected effects on interactions mutations may have. We applied them along with a range of standard bag-of-word features to define nearly 30 features used to build classification models using standard learning algorithms. In the original challenge, the best model provided a roughly 10% (absolute) increase in F1-score as compared to baseline results, based on 10-fold cross-validation in the training data. The benchmarking on the test set shows that our best model achieved higher performance than the baseline model in terms of average precision (~2% higher), recall (~24%) and overall F1 score (4%); in particular, the recall was ranked second over 22 submissions. In post-challenge analysis, we found that relying on standard bioNLP tools to identify entities relevant to PPI affected by mutation relations is inadequate and that the manually defined term lists are effective to produce stronger recall than entity-based methods alone, although this effect was dampened due to variations in the distribution of mutations in the test set as compared to the training set.
For the relation extraction task, we experimented with two methods that leverage the entity recognition and normalization provided by the GNormPlus tool (46). The first method is a method that relies on sentence-level co-occurrence of protein mentions, where only those pairs that are co-mentioned with a frequency (support) above a given threshold are retained. This simple approach was quite effective for the task, given that all documents analyzed could be assumed to describe at least one protein interaction in the context of a mutation. The second method applied supervised machine learning methods to learn the characteristics of protein pairs that are related via the PPI affected by mutations relation; we experimented with SVM graph kernels based on syntactic dependency parses (50) considering both within-sentence and cross-sentential syntactic graphs. These two approaches achieved 26.8% and 28.9% F1 scores, respectively, based on 10-fold cross-validation over the training data. Over the test set, we achieved an F1-score of 34.9% and 35.1% using the co-occurrence strategy and the machine learning method, respectively. Subsequent analysis showed that the main limiting factor for relation extraction performance is in the entity recognition phase; achieving perfect entity recognition can boost the relation extraction performance to an F1-score of nearly 80% and 41% for the co-occurrence and the machine learning approaches, respectively, where the co-occurrence performance is strongly boosted by recall of all relevant entities.
Team 379: Mayo Clinic
We participated in the BioCreative VI Precision Medicine track with one document triage system, and two relation extraction models. Our system, Entity-enhanced Hierarchical Attention Neural Networks (EHANN), is a novel neural network architecture developed for the document triage task. EHANN consists of eight neural network layers, from bottom to top. They are (i) word and entity representation layer, (ii) bidirectional gated recurrent unit (GRU) layer, (iii) attention layer, (iv) sentence and entity-bunch representation layer, (v) bidirectional GRU layer, (vi) attention layer, (vii) document representation layer and (viii) classification layer. First, word and entity sequences are fed into a word and entity representation layer, respectively, and represented by a word or entity annotation. Subsequently, the word and entity level attention layers take word and entity annotations as input to select important words and entities, which are fed into a sentence and entity-bunch representation layer to output sentence and entity-bunch (i.e. a combination of entities from a sentence) representations. Then another attention layer takes the sentence and entity-bunch representations as input to select important sentences or entity-bunches for document classification, and document representations are generated via a document representation layer. Eventually, a softmax function layer is used on the document representation for classification.
The EHANN is an extension to the Hierarchical Attention Network (HAN) (49). HAN includes two attention mechanisms at the word and sentence level so that the model could pay more or less attention to individual words and sentences when constructing the representation of a document. Different from the HAN, the proposed EHANN constructs a document representation by aggregating representations of entities in addition to word and sentence representations as in HAN. This will enable EHANN to especially capture entity relation information beyond word and sentence information. Moreover, the proposed EHANN leverages one more attention mechanism at the entity level in addition to the two attention mechanisms so that various entities are differentially treated.
Team 391: Dalian University of Technology
Our approach for the PPI extraction task can be divided into four steps. Firstly, candidate instances are generated according to the pre-processing method. We select the words between protein pairs and expansion of three words on both sides of protein pairs as candidate instances. Then we extract entity-relation triples from KBs and feed them into TransE model to train the embeddings of entities and relations, namely the knowledge representations. After that, we apply memory network model (MNM) to capture important context clues related to knowledge representations learned from KBs for relation extraction. Finally, the post-processing rules are applied to find additional PPI affected by mutation relations and merge them with the results from MNM. The proposed MNM consists of two memory networks, each of which contains multiple attention layers. The two memory networks share the same set of parameters to learn the weights of the context words between the two entities. To combine the context word and entity embeddings, we do a dimension-wise sum pooling on the attention layer output and entity embedding as the new entity embedding for the next layer. The two final output vectors of the two memory networks and the relation embeddings are concatenated and sent to the softmax layer for relation classification.
We made improvements to our system after the workshop, which can be summarized as follows:
A memory network with four computational layers, with a different attention parameter in each computational layer.
A dimension-wise sum pooling at the end of each layer in memory network, which we compare with dimension-wise max pooling in the experiments.
Additional PPI triples extracted from KBs.
Initializing an entity embedding as the average of its constituting word embeddings, when that protein entity is absent in KB.
Additional post-processing rules.
Based on first four improvements listed above, our system achieves a precision of 40.32%, recall of 32.37% and F1-score of 35.91%. After post-processing, our system achieves a precision of 37.99%, recall of 36.98% and F1-score of 37.48%.
Team 414: Marmara University, Boğaziçi University
We (51) developed three methods for identifying PubMed articles containing genetic mutations affecting PPIs (document triage task). Our first methodology, named Semantic Meaning Classifier with Interaction Network Ontology (SMC-INO), is centered on the idea of meaning computation based on the Helmholtz principle (52, 53). We calculate meaning values for each of the terms from the Interaction Network Ontology (54) present in the documents of each class. The class membership score of a given document is computed by adding the meaning values for all the words in the document for each class. The results showed that this method, the SMC-INO, obtained 52.68% F-score on the Precision Medicine Track test data set. Our second method, called Sprinkled Relevance Value Classifier (S-RVC), is based on the idea of using the most salient terms, generated by Genia Tagger, using the term frequency-relevance frequency metric (55). S-RVC also uses sprinkling (56), which is a process of adding the class labels of documents as additional individual features to the training documents in order to strengthen class-based relationships in the training phase. S-RVC obtained 58.65% F-score on the test data set. The third approach uses a CNN. The CNN model architecture has several layers such as embedding, convolution, max-pooling, dropout and softmax (57). We achieved 66.85% F-score with the CNN model. Moreover, our team also implemented two baseline algorithms and performed a series of experiments on the evaluation data set. According to the experimental results on the evaluation data set, our submitted runs to the shared task achieved higher F-scores than the baseline. The results show promise for the proposed novel techniques, S-RVC and SMC-INO. As expected, CNN demonstrated superiority over the baseline algorithms and future work would lead to further improvements.
Team 418: National Technical University of Athens
The model we proposed for the document triage task is a reusable sequence encoder architecture, which is used as sentence and document encoder. The sequence encoder is a hierarchical bidirectional RNN network equipped with an attention mechanism for identifying the most informative words and sentences in each document. The first level consists of an RNN that operates as a sentence encoder, reading the sequence of words in each sentence and producing a fixed vector representation (sentence vector) as an output from the attention layer. The title sentence vector is separated from the abstract sentences vectors with the later processed by the second RNN layer that operates as a document encoder. Reading the sequence of sentence vectors of the abstract, this layer produces the final vector representation (document vector) at the attention layer. The title’s vector is concatenated with the document vector, and this is used as a feature vector for classification in the last dense layer. With this architecture we achieved 62.89% precision, 76.56% recall and 69.06% F1 score in the document triage task.
Team 419: Taipei Medical University and National Taitung University
We applied two machine learning algorithms to deal with the task of identifying PubMed articles with genetic mutations affecting PPI (document triage task). The first is the support vector machine. We proposed features including n-gram and article-meta information such as MeSH terms and trained two SVM models with the linear kernel. The second is the neural network based on the CNN architecture. We proposed a new CNN structure that integrates convolved context features from different paragraphs and handcrafted features for MeSH term information. The performance of the developed models was evaluated on the training set of the BioCreative VI Precision Medicine document triage task by using 3-fold cross-validation. The SVM-based approach with all developed features achieved the best overall F-score of 68.7%, while the developed CNN model has better precision. We submitted three runs for the test set. The SVM model with all features again achieved the best F-score of 66.9% (recall, 80.7%; precision, 57.2%) while the CNN model has a lower F-score of 60.4% but a higher precision of 59.9%.
Team 420: University of Kentucky
For the end-to-end protein–protein relation extraction task, we employed a three-component pipeline that involves named entity recognition (NER), gene mention normalization and relation classification. For an input article, the NER component is tasked with identifying spans of text corresponding to gene mentions. This is accomplished with the use of a deep neural network designed with character-level CNNs and word-level bidirectional LSTMs such that there is an output layer capable of predicting In, Out, Between (IOB) labels at each timestep. The NER component maps each gene mention to a corresponding gene ID by searching the gene database using the mention itself and additionally cross-referencing the result with a PMID-based query; the latter allows for context-sensitive gene normalization. Lastly, the relation classification component classifies every pair of unique genes in the article as either positive or negative for a PPI relationship. Here we use a standard CNN-based deep neural model for document-level binary relation classification of an entity pair; additionally, ‘entity binding’ is applied wherein participating subject/object pairs are replaced with tokens GENE_A and GENE_B, respectively.
There were several major improvements to the original system. First, we use GNormPlus to augment the original training corpus with additional gene annotations. For the NER component, this has the effect of reducing mixed signals stemming from incomplete gene annotations in the original training data. For the relation classification component, this change allows for the generation of more meaningful negative examples such that the label imbalance more accurately reflects the real-world distribution. Second, during testing, sequences of tokens that are missed by the NER component but appear in the gene lexicon (provided with the BioCreative II Gene Normalization training data) are additionally identified and tagged to enhance overall recall. Lastly, we consult PubTator as a backup source (in addition to results of the PMID-based query) when cross-referencing document-level gene annotations in the gene normalization step. The combination of these changes is responsible for drastically increasing recall while retaining high precision for an overall improved performance of 37.78% micro-F1 (up from 30.03%) on Entrez Gene ID matching and 46.17% micro-F1 (up from 37.27%) on HomoloGene ID matching.
Team 421: Dalian University of Technology
We built a neural network ensemble approach for the BioCreative VI Precision Medicine document triage task (58). In this approach, five individual neural network models [i.e. LSTM, CNN, LSTM-CNN, recurrent CNN (RCNN) and hierarchical LSTM (HieLSTM)] are used for document triage. After post-challenge analysis, to address the problem of the limited size of training set, a PPI pre-trained module with the existing labeled PPI corpora [i.e. BioCreative II (Protein Interaction Article Subtask1, IAS), BioCreative II.5 (Article Classification Task, ACT) and BioCreative III (Article Classification Task-BioCreative III, ACT-BCIII) corpora] is incorporated into each neural network model. Afterwards the ensemble model is built by combining five models’ results via majority voting, weighted majority voting and a logistic regression classification, to further improve the performance. In addition, we explored the effect of additional features (such as part of speech and NER features) to enrich the neural network models in the document triage task.
The experimental results show that (i) our PPI pre-trained module is effective to improve the performances of the deep learning models on the limited labeled PPI affected by mutations data set, and (ii) our ensemble of the neural network models using a logistic regression classification can achieve a further improvement. However, the additional features did not help achieve a further improvement for our ensemble approach in our experiments. Finally, our ensemble achieves the state-of-the-art performance on the BioCreative VI Precision Medicine corpus (71.04% in F-score).
Team 433: Florida State University
For the document triage task, we employed a Gradient Boosted Trees model based on unigrams and bigrams with additional manually engineered features. In addition to unigrams and bigrams, we employed normalized counts of the respective total number of protein names, interaction words and mutation-related words. Protein names were taken from the UniProt database, our dictionary of interaction words was developed in a previous study, while our dictionary of mutation related words was curated from the training data from terms with high term frequency-inverse document frequency (tf-idf) between the relevant and non-relevant labels. Features were also extracted based on a model previously developed by our group for predicting PPI triplets. A triplet consists of two protein names and an interaction word that are all contained in the same sentence. The model is based on features extracted from dependency parses and a set of rules based on grammatical patterns. The predicted probabilities generated by this model were incorporated by taking normalized counts of the number of predicted triplet probabilities lying in equally spaced bins. Features were also extracted directly from the dependency parses of sentences in the abstracts. Shortest paths between key terms in the dependency graphs of sentences were calculated, and the normalized frequency counts of path lengths lying in certain bins as well as bags of words lying along the shortest paths were also extracted and used in the models.
Since the end of the competition, bin sizes for shortest path lengths between key terms and predicted probabilities for PPI word triplets have been selected based upon the distributions seen in the data. Previously, the bin sizes had been chosen arbitrarily. In addition, features based on tf-idf weighted sums of word vectors have been added. These modifications have brought some improvements in performance.
Discussion and conclusions
This community effort was designed to foster development of text-mining tools that while mining scientific literature could collect information of significant practical value in the clinical practice of precision medicine. The success of the precision medicine endeavor depends on the development of comprehensive knowledge base systems that integrate genomic and sequence variation data, with clinical response data, as resources for scientists, health care professionals and patients. Leveraging the information already available in scientific literature, and developing automatic text-mining methods that facilitate the job of database curators to be able to find and curate such valuable information, is the first step toward this goal.
Given the level of participation and team results we conclude that the precision medicine track of BioCreative VI was run successfully and is expected to make significant contributions in this novel challenge of mining PPIs affected by mutations from scientific literature. The training and testing data produced during this effort is novel and substantial in size. Collectively, it consists of 5509 PubMed articles manually annotated for precision medicine relevance. In addition, the corpus annotations include both text spans and normalized concept identifiers for each of the interacting genes in the mutation-affected PPI relations. We believe that such data will be invaluable in fostering the development of text-mining techniques that increase both precision and recall for such tasks. Another important characteristic is that annotated relations in this corpus are at the abstract level because such relations could be expressed across sentence boundaries. The corpus is available from https://biocreative.bioinformatics.udel.edu/tasks/biocreative-vi/track-4/.
Participating teams developed systems that specialized in predicting PubMed articles that contain precision-medicine-relevant information. Curators at molecular interaction databases will benefit from these text-mining systems to select with high-accuracy articles relevant for curation. The top achieved recall was 98%, and the top achieved precision was 62%. Moreover, this is only a first step in this direction. In the future, a system could be built that merges the results of all individual system submissions with high accuracy.
The relation extraction task, on the other hand, showed a somewhat low accuracy. It is to be recognized that this is a very difficult task, and we believe that the accuracy of systems would improve if they were to extract such information from full text. Relation extraction at the abstract level is dependent both on accurate entity recognition and correct normalization, as well as the ability to recognize a relation that spans over sentence boundaries, therefore necessitating a system that goes toward abstract-level understanding.
Acknowledgements
We thank the BioGRID database curators Rose Oughtred, Jennifer Rust, Christie S. Chang and Lorrie Boucher for annotating the evaluation data set.
Funding
National Institutes of Health Intramural Research Program National Library of Medicine; National Institutes of Health Office of Research Infrastructure Programs (R01OD010929 and R24OD011194 to A.C.-a.).
Conflict of interest. None declared.
Database URL:https://biocreative.bioinformatics.udel.edu/tasks/biocreative-vi/track-4/

{kind=link}