





Abstract
Primary amyloidosis of the conjunctiva and eyelid is a rare and often misdiagnosed condition. It is characterized by the deposition of insoluble amyloid fibrils, which are misfolded proteins, in the body. Amyloidosis can be systemic or localized with different types of amyloid fibril proteins identified using mass spectrometry. Ocular involvement in amyloidosis can lead to corneal dystrophies, glaucoma, vitreous opacities, and other symptoms. Diagnosis involves clinical examination and histopathological assessment. Treatment options depend on the extent of involvement and may include surgical excision, glaucoma management, vitrectomy, or liver transplantation in rare cases. We present a rare case of localized conjunctival amyloidosis initially misdiagnosed as pyogenic granuloma, with clinical symptoms of ptosis, periorbital swelling, and conjunctival lesions. The patient underwent excision of the lesions, and subsequent evaluation did not reveal systemic amyloidosis. Ocular amyloidosis requires careful diagnosis and consideration of systemic involvement for appropriate management.
Introduction
Primary amyloidosis of the conjunctiva and eyelid is rare and an often clinically misdiagnosed entity. Amyloidosis is a group of disorders where misfolded proteins form insoluble deposits called amyloid fibrils. These deposits can occur throughout the body (systemic) or in a specific area (localized). There are different types of amyloid fibril proteins, and they can be identified using mass spectrometry. Ocular involvement in the setting of amyloidosis may result in corneal dystrophies, glaucoma, vitreous opacities, among other presentations [1]. Diagnosis involves clinical examination and histopathological assessment. Treatment options depend on the extent of involvement and may include surgical excision, glaucoma management, vitrectomy, or liver transplantation in rare cases [2–5]. We herein present an exceedingly rare case of localized conjunctival amyloidosis clinically presenting with ptosis, periorbital swelling, and conjunctival lesions initially diagnosed as pyogenic granuloma.
Case presentation
This is a case of a 52-years-old male patient who presented to the Ophthalmology clinic complaining of left eye swelling. The left eye had mild ptosis with periorbital swelling and two nodular red–purple soft conjunctival lesions measuring 3–5 mm in size, clinically diagnosed as pyogenic granuloma. There was no associated pain or any other systemic symptoms. CT-scan showed an enhancing soft tissue thickening around the left globe extending from the 9 to 3 O’clock positions. There was no retrobulbar involvement and the extraocular muscles and optic nerve appeared unremarkable. No intraocular abnormalities were demonstrated. In addition, there were no lesions in the brain parenchyma, ventricles, and sinuses. The conjunctival lesions were excised to reveal numerous submucosal interstitial globular deposits of acellular amorphous eosinophilic material. Flow cytometry showed no B-lymphocytes and results were non-contributary. The deposited globular material showed apple green birefringence under polarized light with Congo-Red staining consistent with Amyloid (Fig. 1). Subsequently, the patient underwent a full workup looking for systemic amyloidosis. His Laboratory work up including urine protein electrophoresis, and Immunoglobulin free light chains yielded normal results. Moreover, his complete blood count, liver function test, renal panel, thyroid stimulating hormone (TSH), antinuclear antibody (ANA), and hepatitis panel were all normal. These findings were consistent with localized amyloidosis clinically mimicking pyogenic granuloma. After 6-years, the patient has had no recurrence, no systemic disease, plasma cell dyscrasia, or light chain disease.
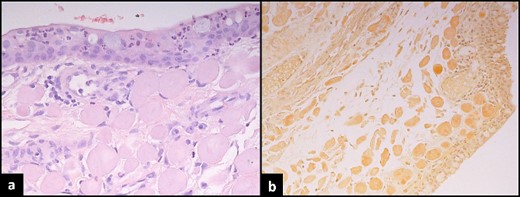
Histopathological assessment of conjunctival lesion. (a) Haematoxylin and eosin stain conjunctival mucosa with underlying globules of cellular eosinophil deposits. (b) Congo-red stain shows apple green birefringence under polarizing microscopy.
Discussion
Amyloidosis is a heterogenous group of disorders characterized by deposition of insoluble amyloid fibrils in extracellular spaces as a result of misfolded proteins. Amyloidosis can either be systemic or localized. In systemic amyloidosis, fibrils are deposited throughout the body, most commonly affecting the kidney and heart. Whereas in localized amyloidosis, deposition of fibrils is limited to a single body site [6]. These proteins have a specific propensity to deposit in connective tissue and perivascular spaces. Alternatively, the terms systemic and localized may refer to the relationship between the site of fibril precursor protein production and the site of amyloid fibril deposition. The Nomenclature Committee of the International Society of Amyloidosis released a revised classification to include four broad groups; primary systemic, primary localized, secondary systemic, and secondary localized. Additional classifications include familial amyloidosis and senile-systemic amyloidosis, otherwise known as wild-type amyloidosis [6, 7].
Amyloid fibrils are synthesized from twisted protofilaments, which are stacks of protein layers in β-sheets warped around each other. Currently, the number of identified types of amyloid fibril proteins is 42, with 24 exclusively in localized disease, 14 exclusively in systemic disease, and 4 which can appear in both. In addition to containing the amyloid fibrils, patients with amyloidosis may also have other signature proteins including serum amyloid P-component and heparan sulfate proteoglycans, apolipoprotein A-IV, and apolipoprotein E, among others. These proteins are considered vital for the stability of the amyloid fibrils and can be identified using mass spectrometry. Amyloid deposits can be classified into immunoglobulin light chains in primary systemic amyloidosis, amyloid A protein in secondary amyloidosis, and Transthyretin in familial amyloidosis [8].
Ocular involvement in cases of amyloidosis has been well established as a potential manifestation of the disease. Amyloidosis has been previously reported in the orbits, extraocular muscles, eyelids, conjunctiva, cornea, iris, and retina. Involvement of the orbits and extraocular muscles typically manifests as unilateral or bilateral tissue infiltration, mass, proptosis, diplopia, epiphora, and foreign body sensation [2]. Both compressive and infiltrative optic neuropathy is exceedingly uncommon, though if present, will manifest chiefly as visual field loss. Amyloidosis of the eyelids and conjunctiva can manifest as ptosis and a yellowish, waxy, typically well circumscribed mass. Corneal involvement results in several corneal dystrophies including gelatinous corneal dystrophy, type 1 and 3 lattice corneal dystrophy, Meretoja syndrome, and type 2 granular corneal dystrophy. Amyloidosis of the lens, iris, and trabecular meshwork may present as secondary glaucoma. Involvement of the vitreous and retina can lead to arteriolar sheathing and non-haemorrhagic vitreous opacities classically described as being ‘glass wool’ in appearance [2, 3].
Diagnosis of ocular amyloidosis can be elucidated clinically by careful history taking, family history, and physical and slit-lamp examination. The use of imaging in the form of a positron emission tomogropahy scan and computed tomography scan (PET-CT), orbital computed tomography scan, optical coherence tomography, and ultrasound biomicroscopy may aid in diagnosis ocular amyloidosis. Additional diagnostic tests may aid in identifying amyloidosis with systemic involvement and include urine and serum monoclonal proteins, bone marrow biopsy, echocardiography, and bone scans. Definitive diagnosis is done by lesion biopsy and careful histologic assessment. Therein, findings such as apple-green birefringence on Congo-red dye, metachromasia with crystal violet dye, and fluorescence under ultraviolet light after thioflavin staining, are all highly diagnostic. [2]
Treatment options for ocular and ocular adnexal amyloidosis depend on the extent of disease involvement and the specific ocular manifestations present. Conjunctival lesions may be managed conservatively, with cryotherapy, or with local excision. [4, 5] Amyloidosis of the lacrimal glands can be managed through surgical excision. Secondary glaucoma in the setting of ocular amyloidosis can be challenging to treat despite maximum topical treatment. Trabeculectomies and shunt placement have been suggested with little report on their long-term success clinically. Vitreous opacities can be managed with pars plana vitrectomy. In the setting of systemic or hereditary disease, liver transplantation may be warranted in order to stop production of amyloid.
Conclusion
We herein present a rare case of ocular amyloidosis with no systemic involvement clinically presenting with ptosis, periorbital swelling, and conjunctival lesions mimicking pyogenic granuloma. Once histologically diagnosed, detailed history, physical examination, and laboratory work-up must be done to detect systemic amyloidosis, if present, for early recognition and appropriate treatment.
Conflict of Interest Statement
The authors declare that there is no conflict of interest regarding the publication of this article. Submitting authors are responsible for coauthors declaring their interests.
Funding
No funding or grant support.
Data availability
The data used to support the findings of this study are included within the article.
Patient consent
Institutional Review Board approval was granted; written consent to publish the case report was waived as this report does not contain any personal information that could lead to the identification of the patient and is sufficiently anonymized according to International Committee of Medical Journal Editors guidelines.
References
Ocular amyloidosis. EyeWiki. (

{kind=link}