





Abstract
A 64-year-old woman presented to the Vascular Outpatient department concerned about a pulsatile swelling in her right supraclavicular fossa. She had no other symptoms. A computed tomography angiogram demonstrated a double aortic arch (DAA) with the innominate artery arising from the right arch and left common carotid and subclavian arteries arising from the left arch. There were no aneurysms. A DAA accounts for 1% of congenital cardiac disease. It is the commonest form of a complete vascular ring, caused by a failure of the embryological, right, fourth pharyngeal arch to regress. Patients typically present in childhood with symptoms arising from tracheal and oesophageal compression, which frequently require surgical intervention. There is a paucity of evidence on how to manage this disease in adulthood, with only a handful of reported cases. Our patient was treated conservatively with advice about potential complications.
INTRODUCTION
This report describes the case of a patient with a double aortic arch (DAA) that presented with a pulsatile neck swelling but no other symptoms. We will outline the underlying pathogenesis behind this rare phenomenon and outline accepted management options for surgical intervention.
CASE REPORT
A 64-year-old woman presented to the Vascular Outpatient department concerned about a 12-month history of an increasingly prominent, pulsatile swelling on the right side of her neck. She was otherwise asymptomatic, with no history of pain, breathing difficulties, swallowing difficulties, cerebral ischaemic events or symptoms of underlying thyroid disease. On examination, she had a 2-cm pulsatile and non-tender swelling in her right supra-clavicular fossa. Her past medical history included type II diabetes mellitus and hypercholesterolaemia. She was normotensive and had no history of cardiovascular disease. Her regular medication was metformin and simvastatin.
She had previously presented in 2002 with a 3 × 4 cm pulsatile mass on the left side of her neck, which was asymptomatic. A duplex scan at the time reported a tortuous aorta with no evidence of an aneurysm. She was subsequently discharged without follow-up.
Figures 1 and 2 are photographs taken of our patient at follow-up.
Patient photograph anterior view.
Patient photograph anterolateral view.
A duplex scan performed on this presentation demonstrated a high, prominent and ectatic aortic arch visible above the sternum, with a diameter of 2.0 cm. The innominate artery appeared ectatic, tortuous with localized wall thickening and had a reported diameter of 1.8 cm. An outpatient computed tomography (CT) angiogram was arranged to help delineate the underlying anatomy. Figure 3 shows a coronal section of this CT angiogram. Figures 4 and 5 show the anterior and posterior views of the 3D reconstruction, respectively.
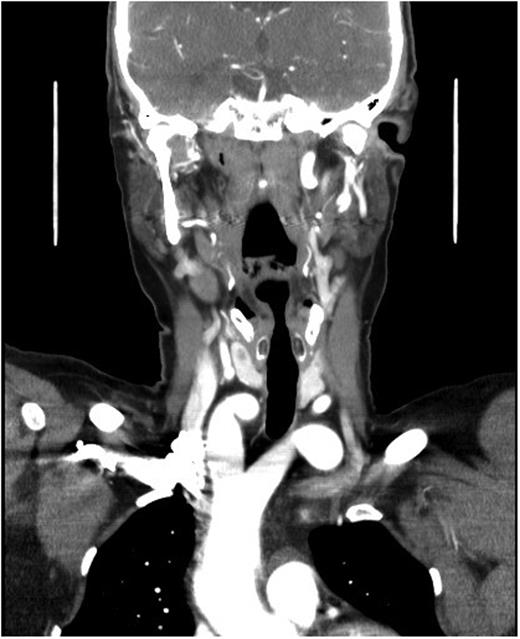
Imaging coronal section.
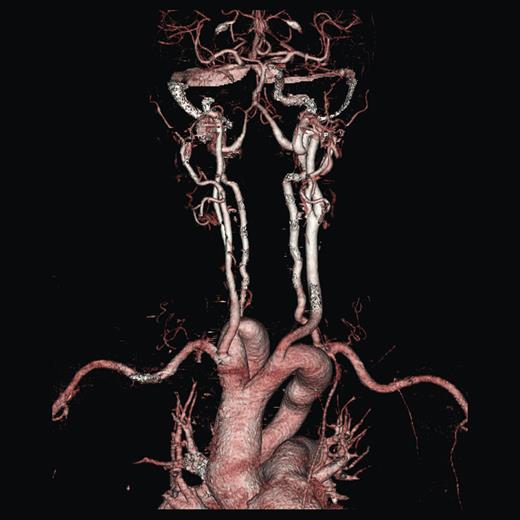
Imaging 3D reconstruction, anterior view.
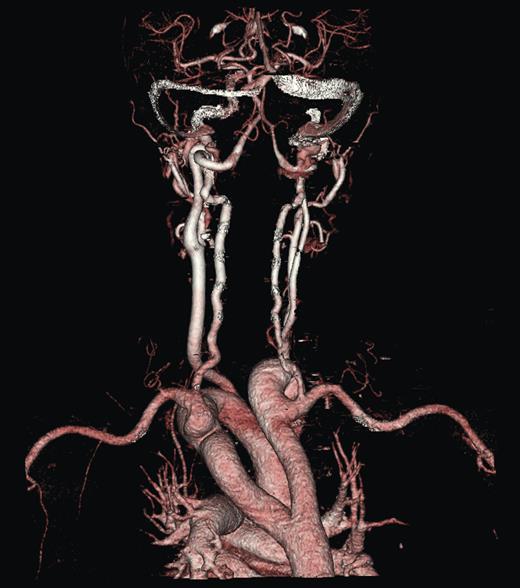
Imaging 3D reconstruction, posterior view.
Figures 3–5 demonstrate a DAA with the right carotid and right subclavian arteries arising from the right-sided arch and left carotid and left subclavian arteries from the left-sided arch. There is also a minor narrowing of the right-sided arch at a kink between the right carotid and subclavian origins. Figure 3 demonstrates both arches rising up into the neck explaining the clinical presentation.
DAA is rare accounting for ∼1% of all congenital cardiac abnormalities, but it is the most prevalent developmental abnormality of the embryological pharyngeal arches [1, 2]. In normal embryological development, the right fourth pharyngeal arch regresses at 36–38 days gestation whilst the left, fourth pharyngeal arch persists eventually becoming the aortic arch. In cases of DAA, the right fourth pharyngeal arch persists forming a right aortic arch [3]. This is in continuity with the left aortic arch dorsally forming a complete vascular ring, which encircles and frequently compresses the trachea and oesophagus.
DAA is commonly associated with other cyanotic congential cardiac abnormalities including Tetrology of Fallot and Transposition of the Great Arteries [1, 4]. McElhinney et al., looking at the frequency of chromosome 22q11 deletions in patients with isolated anomalies of the aortic arch, found chromosome 22q11 deletions in 14% (3 out 22) of patients with a DAA, leading the authors to propose that this genetic abnormality is an important aetiological factor in DAA [5].
Classically, DAA is divided into three main subtypes depending on the dominance and partial atresia of the relative arches. Seventy-five percent of patients with DAA have a right dominant arch, 20% have a left dominant arch with the remaining 5% have a so-called balanced type [6], as was the case with our patient.
Patients with DAA typically present with symptoms resulting from compression of the trachea and oesophagus. Over 90% of patients present with respiratory symptoms, including stridor, dyspnoea, cough and recurrent respiratory infections [1, 7]. Less commonly, patients present with gastrointestinal symptoms such as vomiting and poor oral intake. It is rare for the clinical presentation to manifest outside of infancy. Three separate studies found mean age at presentation to be 1.4 years, 6 and 7 months [1, 7, 8]. There are only a handful (<15) of reported cases of patients presenting in adulthood. These patients have been found to have DAA incidentally during imaging for separate pathology or have had long-standing intractable respiratory/gastrointestinal complaints. As far as we are aware, this is the first reported case of a patient presenting with a pulsatile but otherwise asymptomatic neck lump.
The imaging modality used for identification and delineation of underlying anatomy in DAA varies between studies and reports. Chest X-ray may demonstrate deviation or compression of the trachea, or a right aortic arch contour, however, these findings are frequently absent or may be easily missed. Prior to the advent of CT and magnetic resonance (MR) angiography; barium swallow, echocardiography and duplex imaging were the most commonly used modalities for DAA detection. These modalities, however, are limited by operator/reporter dependency. For an anatomical variant as rare as DAA, many cases may potentially be missed or misinterpreted as in our case. Echocardiography has the advantage of being able to identify intra-cardiac abnormality. Currently, the most robust imaging modalities are CT/MR angiography. Exact anatomy using three-dimensional reconstruction can aid easy interpretation and surgical approach.
The only accepted management for symptomatic DAA is surgical with most centres opting for a left thoracotomy and division of the minor arch. The mean mortality in three separate studies using a left thoracotomy was 1.7% (4/234) [1, 7, 8]. Post-operative complications included bleeding, residual respiratory obstruction, vocal cord paralysis, pneumothorax, pneumonia, chylothorax and feeding difficulties. Prolonged post-operative respiratory complaints have been reported to be as high as 54% [1]. Unfortunately, due to a paucity of evidence, there is no consensus on how to manage asymptomatic patients particularly those that present in adulthood. Subsequently, most reports have advocated a conservative approach [6, 9, 10].
DISCUSSION
In summary, DAA is a rare but important congenital condition, typically presenting in childhood with symptoms arising from tracheal/oesophageal compression. It is exceedingly rare for this condition to go undiagnosed into adulthood with little evidence on how to manage asymptomatic patients. Our patient was managed conservatively with advice about potential complications. The few reported cases in the literature have also chosen this approach.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}