





Abstract
A vast amount of scientific information is encoded in natural language text, and the quantity of such text has become so great that it is no longer economically feasible to have a human as the first step in the search process. Natural language processing and text mining tools have become essential to facilitate the search for and extraction of information from text. This has led to vigorous research efforts to create useful tools and to create humanly labeled text corpora, which can be used to improve such tools. To encourage combining these efforts into larger, more powerful and more capable systems, a common interchange format to represent, store and exchange the data in a simple manner between different language processing systems and text mining tools is highly desirable. Here we propose a simple extensible mark-up language format to share text documents and annotations. The proposed annotation approach allows a large number of different annotations to be represented including sentences, tokens, parts of speech, named entities such as genes or diseases and relationships between named entities. In addition, we provide simple code to hold this data, read it from and write it back to extensible mark-up language files and perform some sample processing. We also describe completed as well as ongoing work to apply the approach in several directions. Code and data are available at http://bioc.sourceforge.net/.
Database URL:http://bioc.sourceforge.net/
Introduction
With the proliferation of natural language text, text mining has emerged as an important research area. As a result many researchers are developing natural language processing (NLP) and information retrieval tools for text mining purposes. However, while the capabilities and the quality of tools continue to grow, it remains challenging to combine these into more complex systems. Every new generation of researchers creates their own software specific to their research, their environment and the format of the data they study; possibly due to the fact that this is the path requiring the least labor. However, with every new cycle restarting in this manner, the sophistication of systems that can be developed is limited.
One bottleneck of text mining research consists of processing data in various formats, writing software to explore data in various formats and implementing algorithms to perform tasks on data in various formats. Typically, the end product of these efforts turns out to be of limited use and not easily adaptable. An interchange data format that can allow the seamless integration of the data in and between many different NLP tools will allow these tools to be leveraged to develop even more impressive and valuable abilities. There are tools that work at the level of the whole document, a section, a paragraph, a sentence, a phrase or just a token. A common format needs to be flexible enough to allow integration of annotations from each of these tools and allowing extension of the text mining infrastructure. For example, the BioCreative protein–protein interaction challenges have addressed document classification, detection of interaction partners, methods of evidence and sentence retrieval. To extend this work, these results all need to be interoperable. Thus to achieve sophistication, we promote reusability. The data reuse problem exists because of the difficulty of achieving interoperability and because of the cognitive burden of learning new systems and languages.
Our goals for this project are simplicity, interoperability, broad use and reuse. We emphasize the simplicity of use in that there should be little investment required to use data provided in a given format or a software module to process that format. Although there is value in the complexity and sophistication of the implementation of an algorithm, there should be no complexity in sharing the results. This will remove the main barrier to reuse of tools and modules, thereby supporting the development of text mining pipelines or systems customized for different workflows.
Not surprisingly, NLP tools need to work and provide value in many and varied environments. Developers use Windows, UNIX, Mac and so forth. Tools may be in C++, Java, Python and so forth. Interoperability requires that data flow in and between these worlds seamlessly. Trade-offs may require that some impressive qualities of a particular platform are not used. With simplicity as a goal, the noteworthy value is that data are accessed in a simple way, and as a result, the same data are more easily treated isomorphically in different languages.
Our approach to these problems is what we would like to call a ‘minimalist’ approach. How ‘little’ can one do to obtain interoperability? We provide an extensible mark-up language (XML) document type definition (DTD) defining ways in which a document can contain text, annotations and relations. Major XML elements may contain ‘infon’ elements, which store key-value pairs with any desired semantic information. We have adapted the term ‘infon’ from the writings of Devlin (1), where it is given the sense of a discrete item of information. An associated ‘key’ file is necessary to define the semantics that appear in tags such as the infon elements. Key files are simple text files where the developer defines the semantics associated with the data. Different corpora or annotation sets sharing the same semantics may reuse an existing key file, thus representing an accepted standard for a particular data type. In addition, key files may describe a new kind of data not seen before. At this point we prescribe no semantic standards. BioC users are encouraged to create their own key files to represent their BioC data collections. In time, we believe, the most useful key files will develop a life of their own, thus providing emerging standards that are naturally adopted by the community.
The XML DTD and the key files are sufficient to provide interoperability, but we take one additional important step. We also minimize the investment needed by a developer to use our approach; we provide data classes to hold documents in memory and connector classes to read/write the XML documents into/out of the data classes. These software classes are provided in C++ and Java. Thus a user of BioC does not have to deal directly with XML and can simply use the already provided classes for reading and writing data.
The details of our approach are laid out as follows: We first discuss related efforts and detail other projects with similar goals. Next, we describe in detail the BioC XML format and how it can be used to share text documents and to allow a large number of different annotations relevant for biomedical research to be represented. We present our data models, discuss implementations and describe working applications. Finally, we conclude with a survey of ongoing and planned projects that have already embraced this initiative, and a description of our vision for further development of these tools.
Related work
A large number of projects have been undertaken with the purpose of enabling or enhancing the prospects for interoperability of software and reusability of software and data. Here we will comment briefly on Text Encoding Initiative (TEI), TIPSTER, Architecture and Tools for Linguistic Analysis Systems (ATLAS), General Architecture for Text Engineering (GATE), Unstructured Information Management Architecture (UIMA) and Linguistic Annotation Framework (LAF) (2–7), and discuss how they relate to our work.
The TEI is a consortium of academic and industrial partners that began in the 1980s and maintains an XML standard for the digital encoding of text in many different genres and forms (http://www.tei-c.org/index.xml). The consortium organizes conferences worldwide, publishes a journal and maintains a Web site with extensive downloadable guidelines, which are currently in version P5. The British National Corpus is available in a TEI compatible format (8) and dictionaries that are a part of the FreeDict Project (http://freedict.org/en/). However, the emphasis of TEI is the humanities, and we are not aware of any text mining efforts that use TEI standards as their basis.
Closer to our interest, many projects have based their efforts to standardize text annotations on TIPSTER (3, 9) and ATLAS (4, 10). The Defense Advanced Research Projects Agency (DARPA) TIPSTER Text program began in 1991 as an effort to develop text retrieval and text mining technologies to enhance US national security, among other goals. Under its auspices the TIPSTER Common Architecture was developed with two of its stated purposes to ‘allow the interchange of modules from different suppliers (“plug and play”)’ and ‘enhance detection and extraction through the exchange of information, and through easier access to linguistic annotations’ (11). The TIPSTER Phase II Architecture Design Document Version 1.52 (3) is a 59-page document describing the object-oriented architecture of a compliant system. It defines an annotation as pertaining to spans of text characterized by integer byte offsets. Thus, it is commonly referred to as the forerunner of standoff annotation. In somewhat later work, Bird and Liberman (10) analysed different approaches to text annotations and concluded that the common element in all of them was the ‘annotation graph’. This led to the ATLAS initiative by the National Institute of Standards and Technology, the Linguistic Data Consortium and the MITRE Corporation with the goal ‘to provide powerful abstractions over annotation tools and formats in order to maximize flexibility and extensibility’. The objective was to allow interoperability based on the ATLAS Interchange Format, an abstract XML representation in standoff form suitable for ‘linear signals (text, speech) indexed by intervals (i.e. annotation graphs), images indexed by bounding boxes, and additional generic representations for other data classes (lexicons, tables, aligned corpora)’. Today TIPSTER and ATLAS are noted as the source of the seminal concepts of the annotation graph and standoff annotations.
The GATE is a Java suite of tools largely developed at the University of Sheffield beginning in 1995. It has roots in the TIPSTER and ATLAS projects and was originally designed as an architecture in which to develop and test new tools and resources for NLP (5). The current GATE Web site (http://gate.ac.uk/) lists eight sources that are suggested as appropriate to cite depending on what resource one may have used in one’s research. An examination of these sources reveals that they, in majority, are contributions from members of the Computer Science Department at the University of Sheffield (http://gate.ac.uk/gate/doc/papers.html). This illustrates that it is hard to get broad support and investment in a system that is designed with the purpose of benefitting the broader community in an important research area. We believe one reason for this may be the ‘perceived’ complexity of the system. GATE user’s guide is a 663-page PDF document that describes a system with an 18-year history of development and elaboration (12). On the other hand, it is important to emphasize that GATE has developed a significant base of software developers, researchers and users in need of text processing tools, with a wide range of interests from scholarly to commercial. They cover a broad variety of text-related systems, and the Sheffield team offers support for addressing a diverse number of language engineering problems.
The UIMA is a software architecture for developing, composing and delivering unstructured information management technologies, which was developed at IBM roughly 10 years ago (6, 13). The intent was to bring efficiencies with a common architecture and uniform data formatting standards for the different teams within IBM working on projects involving NLP. Key elements in UIMA are Text Analysis Engines (TAEs), which are the software modules that perform tagging, parsing, named entity recognition or other NLP tasks. They are required to take in a document in a common analysis structure (CAS) and also produce their output in a CAS. The CAS is an XML and represents the results of processing as standoff annotations related to the TIPSTER and ATLAS approaches as already discussed. In addition to these low level elements, UIMA also instantiates the concept of a collection, a collection reader interface, and collection processing managers to manage the application of particular TAEs or sets of TAEs. UIMA does not prescribe the semantic tags to be used in a CAS implementation, but these need to be appropriately defined to achieve the interoperability and reuse dividends that can be expected from UIMA. UIMA has been embraced at IBM, and one of the positive results has been the IBM Watson question answering system, which won a competition against former Jeopardy stars (14). In 2006, UIMA became freely available through the Apache Software Foundation, and it has been implemented by a number of research teams (15–19). One observation is that different teams implementing UIMA tend to use different semantic tag sets, which creates an interoperability problem between implementations (20). GATE has also wrapped its tools to make them work in a UIMA environment (21).
Since at least 2003, a committee of the International Organization for Standards (ISO/TC 37/SC 4) has been working to develop an LAF that ‘can serve as a basis for harmonizing existing language resources as well as developing new ones’ (7). They have developed a graph-based representation for standoff annotations (22), Graph Annotation Framework (GrAF), which they term a ‘dump format’. A developer is not expected to use this format, but only ensure that what he uses can be mapped isomorphically to a version of the dump format so that through the common dump format his work can become available to others. Portions of the American National Corpus have been annotated in a format consistent with the LAF (23). The committee has also proposed a data category registry (DCR) where data or semantic types as labels or key-value pairs can be registered and become a standard for the field (24, 25). A developer of a resource is expected to use an already defined type set or register a new modified set suitable for the task at hand. We mention this effort because the DCR would be an elegant solution to common types for language processing resources. However, we are not aware that the DCR has received wide use.
In addition to the major initiatives just considered, there have been efforts on a more limited scale to provide annotation standards for particular types of annotations. Much of this has happened as a consequence of providing humanly annotated training data to participants in text mining challenges and workshops, such as BioCreative (26–29), the Joint Workshop on Natural Language Processing in Biomedicine and its Applications (30), the Learning Language in Logic Workshop (LLL05) (31), the BioNLP Shared Tasks (32, 33) and the Collaborative Annotation of a Large Biomedical Corpus Challenge (34–36). Also of note in this context is the work on the Parmenides project (37), the work producing the BioCreative MetaServer, the U-Compare bio-event meta-service (18) and the work of Pyysalo et al. to convert five different annotated datasets of protein–protein interactions to a common format (38). Although these efforts have all been more or less limited in scope and none have led to a widely accepted annotation standard, we see them as important steps in the direction we seek to go and as the natural progenitors of our approach.
There are a number of problems that become evident when surveying efforts to reach interoperability and reusability. First, there is a significant investment in legacy systems that hamper progress. Second, there is no universally accepted standard for tag sets to be used in annotation, and it is difficult to imagine such a standard developing. Different theoretical frameworks, e.g. in dependency parsing, tend to call for different tags (4, 39). Third, much experimental work in NLP is sufficiently different from what already exists as to call for new concepts and new tags and the development of new resources to support the effort (15). The results may never reach the mainstream, and it does not pay to spend a large effort on interoperability and standards compliance until the importance of a new approach becomes clear based on results. Our approach to these problems, a ‘minimalist’ approach, is focused on an XML format, described in a DTD, to share common information. In addition, we have developed a C++ library and Java packages to easily read and write these XML streams. More details and rationales for these choices appear in the remainder of the article.
The BioC workflow allows data in the BioC format, from a file or any other stream, to be read into the BioC data classes via the Input Connector, or written into a new stream via the Output Connector. The Data Processing module stands for any kind of NLP or text mining process that uses this data. Several processing modules may be chained together between input and output.
BioC design
Corresponding to the objectives of the BioC initiative, the BioC design envisions a simple workflow for the many different NLP and text processing tasks. In this workflow, shown in Figure 1, first the data prepared in the BioC format is read into the BioC data classes via an Input Connector. The XML input may come from a file or a network stream such as a web server or client. Next, the Data Processing module stands for any kind of NLP or text mining process that is desired to be performed on this data. Because compliant data modules use BioC data classes for input/output, several processing modules may be chained together without additional XML input/output. When the final desired output is achieved, the BioC class containing the output data is passed to an Output Connector and a new data file is produced in the BioC format, ready to share with the community and be reused for other purposes or applications. BioC code may be convenient, but not necessary for internal data processing. The BioC design allows flexibility and the three main components Data Input, Data Processing and Data Output, can be decoupled at any time. In this section, we describe these modules in detail, in particular our choice of XML as the basis of the BioC data exchange and the BioC data classes. Currently BioC is implemented in Java and C++.
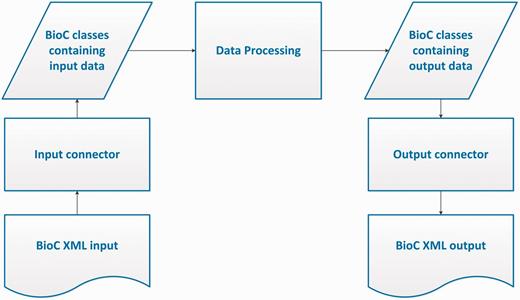
BioC process sequence.
The BioC data model
A flexible data model needs to fulfill these requirements: it is easily represented in common languages, it is easily recorded in a well-known file format and it is portable between different operating systems and environments. Describing a data model using an XML DTD avoids leaning on implementation language features and provides a standardized file format, familiar to researchers from different backgrounds. In addition, libraries to read and write XML files are available for most computer languages and systems. Another possible tool for describing biomedical text and annotations would be the Resource Description Framework (RDF), a standard model for data interchange on the Web (http://www.w3.org/RDF/). Although the ecosystem surrounding RDF, such as OWL and SPARQL, is intriguing, XML is better known, more widely used and adequate for our purposes. In addition, many biomedical resources, such as clinical data, may never be directly available on the Web. Nonetheless, several of us are investigating the best way to combine benefits of RDF and OWL with BioC.
Similar to other possible formats, XML has both advantages and disadvantages. In particular, it is a verbose format. However, it is well known, well documented and well implemented. XML allows the file structure to be precisely and unambiguously described in DTDs. In addition to providing guidance to human developers, an XML file can be validated against a DTD in which case it is guaranteed to work with any software that handles files matching that DTD. The BioC XML DTD is shown in Figure 2 and is available on a specific URL so it can be directly accessed. The elements are described in Table 1 and discussed below.

BioC.dtd.
Elements in the BioC.dtd
| Element | Description |
|---|---|
| Collection | A group of documents, usually from a known corpus. |
| Source | Name of the corpus or other source where the documents were obtained. |
| Key | Reference to a separate document describing the details of the BioC XML file. It should include all information needed to interpret the data in the file such as types used to describe passages and annotations. For example, if a file includes part-of-speech tags, this file should describe the part-of-speech tags used. An HTML URL would also be a useful way to reference a key file. |
| Date | Date when the documents were extracted from the original corpus. It may be as simple as YYYYMMDD, but any reasonable format described in the key file is acceptable. |
| Infon | Key-value pairs can record essentially arbitrary information. |
| Attribute: | |
| Key: it is assumed to be unique within each element. | |
| For example: key = ‘type’ will be particularly common. For PubMed documents, passage ‘type’ might signal ‘title’ or ‘abstract’. For annotation elements, it might indicate ‘noun phrase’, ‘gene’ or ‘disease’. The semantics encoded in the infon key-value pairs should be described in the key file. | |
| Document | A document in the collection. A single, complete and stand-alone document. |
| id | id of the document in the parent corpus. Should be unique in the collection. |
| Passage | One portion of the document. PubMed documents have a title and an abstract. Structured abstracts could have additional passages. For full-text documents, passages could be sections such as Introduction, Materials and Methods or Conclusion. Another option would be paragraphs. Passages impose a linear structure on the document. |
| Offset | Where the element occurs in the parent document. They should be sequential, avoid overlap and identify an element's position in the document. An element’s position is specified with respect to the whole document and not relative to its parent element’s position. |
| Text | The original text of the element. |
| Sentence | One sentence of the passage. |
| Annotation | Stand-off annotation. |
| Attribute: | |
| id: referred to by relations. | |
| Location | Location of the annotated text. Multiple locations indicate a multispan annotation. |
| Attributes: | |
| offset: document offset to where the annotated text begins in the sentence or passage. | |
| length: byte length of the annotated text. | |
| Relation | Relation between annotations and/or other relations. |
| Attribute: | |
| id: referred to by other relations. | |
| Node | The annotations and/or other relations in this relation. |
| Attributes: | |
| refid: id of an annotation or other relation. | |
| role: describes how the referenced annotation or other relation participates in the current relation. |
Although a DTD file describes the structure of an XML file, additional information, such as the data semantics, must be known before the data in the XML file can be effectively used. We put this information in a key file that accompanies any BioC XML file. The key file allows the creator to specify details of how the data in the XML file should be interpreted, and what assumptions were made when preparing the data. In principle, this allows a lot of flexibility for data representation in the BioC XML file.
We do not prescribe all the higher level semantics of the information stored in the BioC data model. If two different gene annotations use different semantics to describe the annotations, changing between the two will be difficult. If two projects use distinct sets of specifications, it may be difficult for them to harmonize the semantics. The BioC initiative aims to facilitate interoperability when these other questions have been addressed.
We recommend that the community adopt certain key files as best practices. For example, a BioC XML file containing part-of-speech annotations would be most useful if it followed a community accepted part-of-speech key file based on a widely used set of part-of-speech tags. Success will depend on creating widely used key files that are appropriate and adequate for commonly used data types.
An important feature of the BioC XML format is the ‘infon’ element, which stores a key-value pair with any desired semantic information. Key files should define the possible ‘key’ strings and describe possible ‘value’ strings. Because infons appear within different elements, the level of information that they carry will depend on the context. A passage infon with key=‘type’ may signal different sections in the full text document, with values such as ‘Introduction’ or ‘Methods’. On the other hand, an annotation infon with key=‘type’ may indicate ‘gene’, ‘disease’ or ‘biological event’. Even more specific, in a disease concept corpus, annotation infons where value strings are MeSH concept IDs of the annotated disease strings will have key=‘MeSH’, and infons where value strings are SNOMED CT concept IDs will be paired with key=‘SNOMED’. These possibilities must be sufficiently covered in the key file accompanying a BioC corpus. Elements of the BioC XML are detailed in Table 1, and we have included several key file examples in the Supplementary Material to illustrate more variations of the BioC data.
As detailed above, the BioC data model is capable of representing a broad range of data elements from a collection of documents through passages, sentences, down to annotations on individual tokens and relations between them. Thus it is suitable for reflecting information at different levels and is appropriate for a wide range of common tasks. Ide and Suderman (40) argue that GrAF is coextensive with UIMA and GATE in what it can represent. GrAF is based on a graph structure, and BioC using relations can easily represent a graph. Therefore, we argue that, for textual data, BioC can represent these same structures. However, a mapping from BioC to GrAF is not available at this time. As with any approach, BioC has limitations. It targets tool developers and not end users, and it focuses on text data rather than other media. Even when BioC can represent a particular kind of data, it may not be the most convenient way to represent that data. For example, if one wishes to represent a graph structure, GrAF may be more convenient, or if one needs to represent a system of type priorities, UIMA may be more convenient.
In the rest of this section we use a running example to illustrate the BioC file format. The running example is an arbitrary excerpt from a PubMed Central® (PMC) article (PMC3048155). It illustrates the different levels of BioC data, including a data collection, a document in the collection, passage and sentence segmentation, annotations and relations. We describe the XML elements in Table 1 and give excerpts of possible BioC XML files in Figures 3–6. See the Supplementary Material for the implementation in C++ and Java: the data classes to hold documents in memory and connector classes to read and write the XML documents.

The exampleCollection.xml.

The exampleCollection.key file describing the elements of the exampleCollection.xml file.
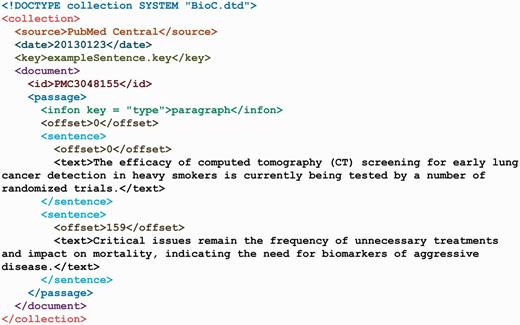
The exampleSentence.xml.

The exampleAnnotation.xml.
Collection of documents
The most fundamental data for NLP is a collection of documents. This is the starting point for the BioC XML file format. Indeed, a collection is just a series of documents to which we add information about the original source and the time the collection was created. Documents may be simple, or they may contain a lot of internal structure. Explicitly capturing all that structure in the XML would require unwanted complexity. In the BioC XML format, documents consist of a series of passages. Introduction, Methods, Results and other sections defined in a journal article may be treated as passages. If desired, a complete outline structure could be duplicated by appropriate key-value pairs in infon elements. But the document could still be simply processed as a list of passages. An example of this information is depicted in exampleCollection.xml (Figure 3) and in exampleCollection.key (Figure 4).
Sentence segmentation
Sentences are an important feature of text documents, and their distinction is important for many NLP tools. The BioC XML format has an option for them to be explicitly marked. Each passage can contain a series of sentences instead of the text of the passage. The XML file illustrating this is shown in Figure 5. A sentence’s offset is specified with respect to the whole document and not relative to the offset of the passage it is in. This ensures consistent references to the original text.
Text annotations
Much of the input and output for biomedical text processing programs can be expressed as annotations to the surface text. Annotations can represent anything, whether convenient and simple, or not. Examples include linguistic features such as tokens, part-of-speech tags and noun phrases. Biomedical examples include genes, diseases and parts of the body. The location tag connects this information with the original text. To promote modular reuse of the data, we recommend that different annotation types are stored in different BioC files, but this is not a requirement. An annotation is typically a single continuous segment of text, but multi-segment annotations are also allowed. Because annotations are standoff they may be nested or overlapping. An annotation example is shown in Figure 6. In this case, the annotation appears within a particular sentence. For flexibility, the BioC DTD allows annotations to appear directly in each passage. This provides for NLP tasks not limited to a single sentence. Note the optional, but recommended, attribute ‘id’. This allows the annotation to be referenced in relations.
The location elements include an offset attribute for the document offset to where the annotated text begins, and a length attribute for the length of the annotated text segment. Multiple location elements allow for multi-segmented annotations. For example, in the text ‘red and white blood cells’, both ‘white blood cells’ and ‘red blood cells’ should be annotated.
Table 2 provides a sampling of different annotations that can be easily represented in the BioC format, including a multi-segmented annotation example, using only the first sentence of the running example.
Possible annotations in the BioC format
| id | Infon Key: value | Location | Text | Comments | |
|---|---|---|---|---|---|
| Offset | Length | ||||
| T4 | Part of speech: NN | 25 | 10 | Tomography | Part of speech tagging |
| L14 | Lemma: smoker | 92 | 7 | Smokers | Lemmatization of token |
| A1 | ABRV: Long Form | 16 | 19 | Computed tomography | Abbreviation (ABRV) definition in text |
| A2 | ABRV: Short Form | 37 | 2 | CT | Abbreviation in text |
| D1 | Type: disease | 61 | 11 | Lung cancer | Disease name mention in text. |
| D1 | MeSH: D008175 | Concept in terminology resource | |||
| E1 | Type: event | 16 | 19 | Computed tomography screening | Segmented mention annotation |
| 41 | 9 | ||||
The efficacy of computed tomography (CT) screening for early lung cancer detection in heavy smokers is currently being tested by a number of randomized trials.
Relation annotations
Just recognizing named entities and other textual features is no longer sufficient. Biomedical text mining research has progressed to detect and report relations between these elements. Examples include protein–protein interactions, gene–disease correlations and so forth. To describe a relation, one needs to specify a list of annotations or relations that participate in the relation and roles for how each item participates in the relation. Again, an ‘id’ attribute allows a relation to participate as a member of other relations. Annotations can appear at the passage or sentence level. Relations can appear at the document, passage or sentence level.
For a BioC relation example, consider the annotations listed in Table 2. A relation can be defined between the Long Form: computed tomography and Short Form: CT pair, to express that these two strings define an abbreviation in text, as shown:
<relation id="R1">
<node refid="A1” role="Long Form"/>
<node refid="A2” role="Short Form"/>
</relation>
The BioC DTD has been used to express complex relations including dependency parses, full syntactic parse trees and the BioNLP shared task data annotations (http://www.nactem.ac.uk/tsujii/GENIA/SharedTask/). Another relation example depicting nested protein–protein interaction events can be found in the Supplementary Material.
BioC data model in biomedical text mining research
Although this is the first formally published description of BioC, BioC is already being used in the BioNLP research community. For example, the BioNLP2013 Shared Task (http://2013.bionlp-st.org/), which was completed in April 2013, listed their corpus data and useful annotations in the BioC XML format as supporting resources for all participating teams, publically available for anyone to download (http://2013.bionlp-st.org/supporting-resources). In addition, the BioCreative IV challenge (http://www.biocreative.org/events/biocreative-iv/CFP/), scheduled to take place in October 2013, consists of five distinct tasks. Of these, Track 1, Interoperability, is dedicated to BioC; Track 3, Comparative Toxicogenomics Database Curation, and Track 4, Gene Ontology (GO) curation, have adopted BioC as their sole data format; and finally, Track 5, Interactive Curation, strongly encourages all participating teams to use the BioC XML format.
BioNLP community events are usually organized around specific biomedically relevant tasks of information extraction and are provided significant sized corpora of journal articles. For example, the BioNLP’09 Shared Task (http://www.nactem.ac.uk/tsujii/GENIA/SharedTask/) (41) included data annotations for proteins, protein–protein interaction events and modifiers of those events in >1200 articles. The challenge organizers have come up with a specialized format to describe this data. We were able to easily use the general-purpose BioC DTD to express all these complex nested relations. We have also repeated this exercise with the BioNLP’11 Shared Task (https://sites.google.com/site/bionlpst/) dataset (33) and BioNLP’13 Shared Task (http://2013.bionlp-st.org/) datasets. These latter tasks expanded the number of annotated events, increased the size of the datasets and added more information extraction and recognition tasks related to those corpora.
The first steps in any text mining task usually involve basic steps such as sentence segmenting, tokenization, lemmatization and part-of-speech tagging. To address such processing, we converted MedPost (42) and BioLemmatizer (43) into BioC-compliant tools that read and produce their output in the BioC format. On a more specialized level, it is often useful to detect abbreviation definitions in medical text before attempting higher level entity recognition tasks such as disease and gene/protein recognition, as these often appear in an abbreviated form. To address this, we have again produced BioC-compliant versions of the abbreviation definition detection tools of Sohn et al. (44) and Shwartz and Hearst (45). This is just the beginning of an ongoing project to make more tools and corpora available to the community. Implementation details, further discussion and downloads can be accessed on the BioC Web site.
Current BioC work
Computational biology research is integrally dependent on the accuracy of NLP and text mining tools for purposes of information retrieval and extraction. However, as discussed in the Related Work section, these services are dispersed, may include proprietary software and are often integrated in specific packages imposing considerable overhead. To promote progress in the field, it is important to facilitate better access to the tools, methods and, in particular, data and the produced results.
The BioC project is supported by a number of prominent researchers in the biomedical text mining field with an interest in the BioCreative challenge evaluations and workshops (26–29). Concept, design, data, code and documentation were shared from the early stages of the initiative. In this section, we provide our views on the utility of the BioC proposal and where to go next. Having a common interest of progressing toward more complex biologically relevant research problems, it is important that we are able to provide carefully prepared training and test data collections, and tools to access them, to facilitate research.
Don Comeau and Rezarta Islamaj Doğan
Don Comeau and Rezarta Islamaj Doğan are Staff Scientists at the National Center for Biotechnology Information whose experience and research cover many aspects of information extraction and text mining for biomedical literature.
We believe that the BioC initiative will be most useful in facilitating data exchange between research groups and developing accompanying programs that will facilitate its use and reuse. Having simplicity in mind as our fundamental principle, we are preparing for general release the whole open access PMC set of full text articles in the BioC XML format. This set of articles, although available for download from the PMC Web site, is not convenient for text mining research. The PMC XML data model, designed to preserve all original article details without loss, incorporates great flexibility to meet the organization and display needs of many different publishers. The release of the PMC open access corpus in the BioC XML format is important because it will provide a large scale corpus of full text articles in the biomedical domain, fully and freely available for biomedical research in an easy-to-use format for text processing applications.
Next, we target the most common tasks where we think reuse is a meaningful expectation. To pursue this goal, we will release a suite of basic NLP methods that can be used with BioC-formatted input data, such as sentence and token segmenters, part-of-speech tagging with MedPost and Stanford part-of-speech taggers, abbreviation definition detection in PubMed articles with several algorithms (44–46) and so forth. This suite slightly modifies the original works to make them compatible with the BioC XML input format and produces the output data in the BioC XML format.
Paolo Ciccarese
Paolo Ciccarese is a senior researcher at Massachusetts General Hospital and Harvard Medical School. Paolo is a co-chair of the W3C Open Annotation Community group and the principal software architect of the open-source Domeo web annotation toolkit.
Domeo (47) is an extensible web application that enables users to efficiently create, curate, refine and share free-form and ontology-based annotations on online textual documents. Domeo supports manual, semi-automated and fully automated annotation with complete provenance records and supports multiple repositories with peer-to-peer sharing. The annotation product is currently shared in Annotation Ontology (48) RDF format, and Domeo is being extended to support the Open Annotation format as well (http://www.openannotation.org/spec/core/).
Domeo has also been designed to leverage text mining algorithms made available through external web services. Those results can be displayed in the Domeo user interface, which provides tools for curation of annotation results preserving data provenance. This curation can be part of the display of the document presentation and can help the text mining providers to improve the performance of their services.
Currently, integration of Domeo with text mining services is performed both through ad hoc and through standardized software components able to translate results into the Annotation Ontology format. The modules developed for the Apache Clerezza Project allow for an automatic translation of the results produced using the UIMA text mining framework into the Annotation Ontology format. We plan to extend Domeo to support the BioC data exchange format, and we also plan to work with the W3C Open Annotation Community Group to convert the BioC content into RDF content in compliance with the Open Annotation Model.
Martin Krallinger, Florian Leitner and Alfonso Valencia
Martin Krallinger and Florian Leitner are research scientists at the Structural Computational Biology Group of the Spanish National Cancer Research Centre led by Alfonso Valencia. Their main research interests are related to text mining, information extraction and retrieval applied to biomedical and molecular biology literature. They have co-organized several BioCreative text mining challenges and evaluation tasks.
Annotated biomedical corpora created for community challenges are among the most heavily used resources for the implementation of new biomedical NLP applications. These corpora serve to evaluate the performance of heterogeneous systems on a common task and data collection. Being able to ‘align’ and visualize annotations from different tools in a single format is a challenging mission and was attempted initially by the BioCreative metaserver platform for a limited set of annotation types (49). Unfortunately such evaluation corpora were distributed to the community in a range of different formats that supposes a considerable workload for participating teams to adapt their methods to a particular task, being thus one of the factors influencing the dropout rate of registered participants. Corpus refactoring, i.e. changing the format of a corpus without changing its underlying semantics, can help to increase its usage (50). We foresee that the use of the BioC XML format, as a common data annotation format, might lower the adaptation burden for text mining developers on one side and also facilitate that system developers make reuse of community challenge corpora after the official competitions are over on the other side. Our research group will explore the adaptation of two data collections to the BioC XML format and the integration of this format within the UIMA framework. The first dataset consists in the Alzheimer’s Disease Literature Corpus that was used for a task dealing with ‘Machine reading of biomedical texts about Alzheimer’s disease’, posed at the Question Answering for Machine Reading Evaluation (QA4MRE 2012). A total of seven teams participated in this task with the goal of applying machine reading systems to answer questions about Alzheimer’s disease. The second document collection whose adaptation to the BioC format will be examined is the dataset currently prepared for the CHEMDNER task of BioCreative IV on chemical compound and drug name recognition from text (http://www.biocreative.org/tasks/biocreative-iv/chemdner/). This set consists of annotated mentions of chemical compounds and drugs in text, designed for a classical named entity recognition task. Despite the declared goal of the BioC standard to eliminate the requirement of complex frameworks for enabling interoperability, this format nonetheless can be used in more complex environments as well. Therefore, we are investigating the design of UIMA collection readers and CAS consumers tailored to the BioC standard. This way, we would provide access to the BioC interoperability format via this framework. To make this application interface independent of any underlying UIMA type system, an approach similar to the design implemented by the OpenNLP Annotation Engine wrappers could be applied to provide UIMA handlers that encapsulate the already existing BioC XML Java parsers and producers.
Zhiyong Lu
Zhiyong Lu is an Earl Stadtman Investigator at the National Center for Biotechnology Information (NCBI). He is one of the organizers of the BioCreative challenges, and his research group has worked on a wide range of text analysis problems, from biomedical data curation to drug repositioning.
We envision several efforts to make use of BioC in real-world applications. First, we plan to release the newly developed National Center for Biotechnology Information disease corpus (51) for download in the BioC XML format in addition to the current tab-delimited format. Second, in the 2013 BioCreative IV GO task (http://www.biocreative.org/tasks/biocreative-iv/track-4-GO/), a challenge event for tackling a major bottleneck in biocuration (52), we plan to use the BioC standard to prepare the training and test data that consist of both full-length articles in PMC and associated human annotations (GO terms with evidence sentences). Finally, we would like to apply BioC to several software tools developed for biomedical named entity recognition and normalization such as SR4GN (53) and BANNER (54). We believe these efforts will lead to improved interoperability of these resources and tools, thus making them more valuable to the text mining research community and beyond.
Yifan Peng, Manabu Torii and Cathy Wu
Yifan Peng is a doctoral student and Manabu Torii is a research assistant professor at the University of Delaware Center for Bioinformatics & Computational Biology, directed by Cathy Wu. The center has developed a number of text mining systems and resources (55–57) and coordinated community efforts for biological text mining (28, 29).
While aiming for full adoption of BioC for broad dissemination of the text mining resources developed at the University of Delaware center (http://www.proteininformationresource.org/iprolink/), including curated literature corpora and text mining tools, our demonstrative project for BioC is a text mining module for sentence simplification that can be reusable in various workflows or systems. The sentence simplification module named iSimp (58) produces one or more simple sentences from a given sentence by reducing its syntactic complexity (http://research.bioinformatics.udel.edu/isimp/). For example, given a complex sentence such as ‘Active Raf-2 phosphorylates and activates MEK1, which phosphorylates and activates the MAP kinases signal regulated kinases, ERK1 and ERK2, (PMID-8557975)’ iSimp produces multiple simple sentences, including ‘Active Raf-2 phosphorylates MEK1’, ‘MEK1 phosphorylates ERK1, ’MEK1 activates ERK1’ and so forth. The underlying assumption is that this simplification can improve the performance of existing text mining applications. However, sentence simplification is different from most NLP tasks in that it not only annotates input text, but also generates new sentences. To make iSimp readily adaptable for various applications in the biomedical domain, we adopt the BioC because it allows us to define and embed both original and generated sentences using a simple standard format. With its simplicity and flexibility, the BioC framework would ease the incorporation of iSimp results into a text mining pipeline.
Fabio Rinaldi
Fabio Rinaldi is a senior researcher at the University of Zurich. He is the leader of the OntoGene group and principal investigator of the Semi-Automated Semantic Enrichment of the Biomedical Literature project.
OntoGene (www.ontogene.org) is a research project focused on the extraction of semantic relations between specific biological entities (such as genes, proteins, drugs and diseases) from the biomedical scientific literature. As such, the OntoGene team has developed several successful biomedical text mining applications, centered on an XML-based pipeline, that have been tested in community-wide competitions, with top-ranked achievements (59, 60). The OntoGene system is used to generate annotations that can be accessed and modified through the OntoGene Document Inspector interface. The system aims to facilitate the work of database curators and increase their work efficiency through a process of assisted curation. The high usability of OntoGene Document Inspector was the question of an experiment performed in collaboration with the PharmGKB group at Stanford University (61).
The OntoGene system relies internally on an XML-based document representation with similarities to the proposed BioC format. We plan to adapt some core components of the pipeline to make them capable of seamlessly handling the BioC format. We also plan to provide XSLT-based (http://www.w3schools.com/xsl/) converters to map the current OntoGene format to the BioC format and in the opposite direction. We strongly believe that a common format for different levels of the annotation process will enhance the utility of text mining tools and allow speedier progress in the field.
Karin Verspoor
Karin Verspoor is a senior researcher and leader of the Biomedical Informatics team at the National ICT Australia (NICTA) Victoria Research Laboratory. She is a computational linguist with research interests focused on information extraction and text mining applications in the biomedical domain.
Within the semantic web community, there are currently ongoing efforts to address standardization of annotations, including but not limited to linguistic annotations (62, 63), over web resources, including documents and other media resources. One important effort along these lines is the work of the W3C Community Group on Open Annotation (http://www.w3.org/community/openannotation/). These efforts address the same fundamental goal as BioC, to enable interoperability of annotations over resources. Although the target audience of the W3C for the representation may vary, and the tools available to work with data representation are not designed specifically for the NLP community, some level of alignment could benefit both research efforts. In particular, we identify the emphasis on well-defined semantic types, provided as common URLs and defined through ontological specifications as a relevant element. Previous work has pointed out the advantages in the UIMA context of reuse of external type or concept identifiers (20). Therefore, we intend to explore synergies between the BioC framework and other semantic web efforts, with the aim of building tools that convert between BioC and RDF-based representations, to enable broader reuse of BioC annotated content.
Thomas C. Wiegers
Thomas C. Wiegers is a research bioinformatician in the Department of Biology at North Carolina State University. He is one of the organizers of the BioCreative challenges, and among his research focus areas is the text mining pipeline at the Comparative Toxicogenomics Database (CTD) project.
The CTD (http://ctdbase.org) is a publicly available resource that seeks to elucidate the mechanisms by which drugs and environmental chemicals influence the function of biological processes and human health (64, 65). The CTD curators manually curate peer-reviewed scientific articles to identify chemical–gene/protein interactions, chemical–disease relationships and gene–disease relationships (66). The CTD staff organized the BioCreative 2012 Track I Triage task (67), which focused on developing tools that ranked articles in terms of their curation potential, and also identified gene, chemical and disease names per article. In retrospect, the tools built as the result of the workshop, although impressive, would have been more valuable to CTD had they been built with interoperability in mind. The tools developed by participants were written using a wide variety of technologies and within technical infrastructures that would not necessarily easily integrate directly into CTD’s existing text mining pipeline. In short, interoperability was a major impediment to the direct application of the collaboration to the CTD pipeline. CTD is now organizing a track for BioCreative IV, with a focus on interoperability (http://www.biocreative.org/tasks/biocreative-iv/track-3-CTD/). We ask participants to build interoperable tools that can be accessed remotely by batch-oriented CTD text mining processes via web services, using BioC as the sole communications interchange framework. With this track we wish to examine and resolve several questions, e.g. can CTD, using technologies such as web services, directly integrate text mining tools running on remote platforms? Can BioC be used as the basis for communication exchange between these remote platforms? Would the response time associated with such an architecture be suitable for asynchronous batch processing-based text mining? This web services-based approach to text mining, if successful, could serve as a proof-of-concept to decouple the potentially disparate technical infrastructures of text mining integrators and their service providers, and standardize communication interchange across dispersed research groups.
Conclusion
We have described the BioC format that can be used to exchange prepared biomedical corpora and any accompanying annotations between different research groups and software platforms. This interchange data format will increase cooperation and allow construction of more powerful and capable systems. We have also made available data classes to hold documents in memory and connector classes to read/write the BioC XML documents into/out of the data classes. These software classes are currently provided in C++ and Java and are planned for other languages as well. Thus a user of BioC does not have to deal directly with XML and can simply use the already provided classes to read and write the data. More details, data and source code can be found at the project webpage (http://bioc.sourceforge.net/).
The proposed BioC framework invites a variety of applications. For example:
Creating a corpus of annotated data in BioC format;
Taking open-source data that are available and converting it to BioC format;
Developing a tool to map a common data format to BioC format and vice-versa, so that those who have access to the data may use the tool to produce the BioC format for further processing;
Designing a BioC-compliant annotation tool that lets the user create an annotation output in BioC format;
Taking an existing NLP or Bio-NLP tool and converting it to a BioC-compliant tool;
Creating a new BioC-compliant NLP or Bio-NLP tool.
The current BioC contributions touch on a variety of issues, areas for improvement and further development of text mining tools for better access and understanding of the biological literature. The BioC interoperability initiative is organized as a track in BioCreative IV (http://www.biocreative.org/tasks/biocreative-iv/track-1-interoperability/), serving as a foundation for other BioCreative IV tasks. The findings from these tasks will provide insights in real-world applications and further identify functional requirements and community needs for future development.
The ultimate goal motivating the BioC undertaking is to create a common platform to facilitate data exchange and data and tool reuse. With the efforts outlined earlier, we believe that the applicability of text mining tools will broaden, their performance will improve and the use and reuse of biomedical corpora will increase.
Acknowledgement
The authors thank the anonymous reviewers for their useful suggestions in improving this manuscript.
Funding
Intramural Research Program of the National Institutes of Health, National Library of Medicine to D.C.C., R.I.D., Z.L. and W.J.W; National Library of Medicine [G08LM010720] to C.H.W.; National Institutes of Health [NIH 5R01 LM009254-07, NIH 5R01 LM008111-08] to K.B.C.; National Science Foundation [DBI-1062520] to Y.P., M.T. and C.H.W.; and Swiss National Science Foundation [105315_130558/1] to F.R.
Conflict of interest. None declared.
References
Author notes
Citation details: Comeau D.C., Doğan R.I., Ciccarese P., et al. BioC: a minimalist approach to interoperability for biomedical text processing. Database, (2013) Vol. 2013: article ID bat064; doi:10.1093/database/bat064.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}