





Abstract
Adrenal incidentalomas originally defined as tumors discovered serendipitously in the course of diagnostic evaluation or follow-up of unrelated disorders, may occasionally pose serious diagnostic challenges. Intravascular large B-cell lymphoma (IVLBCL) may be a rare example of such a case. We present an IVLBCL confined to the adrenal gland in a 52-year-old man focusing on its diagnostic and therapeutic aspects. On endocrine work up, the tumor was hormonally inactive and exhibited inconclusive imaging characteristics without signs of locoregional spread. After a left laparoscopic adrenalectomy, histologic sections revealed the presence of tumor cells inside dilated, thin-walled vascular spaces. Immunohistochemical stains confirmed the diagnosis of IVLBCL. The patient was then referred to a Hematology Unit for further staging and treatment and received six cycles of R-CHOP. Despite the fact that IVLBCL carries a dismal prognosis our patient remains alive and in complete remission 6 years after the initial diagnosis.
INTRODUCTION
Intravascular B-cell lymphoma (IVLBCL) is a rare, usually lethal type of non-Hodgkin lymphoma characterized by selective growth of lymphoma cells within the lumen of vessels [1]. Adrenal involvement is exceptionally rare and may occur either unilateral or bilateral. We describe herein a case of an isolated adrenal IVLBCL in a 52-year-old man and discuss its clinical presentation, diagnosis and implications of treatment.
CASE REPORT
A 52-year-old male with an unremarkable past medical history was referred to our Clinic due to an isolated left adrenal mass and complains of recent onset of proximal muscle weakness. Routine hematologic and biochemical indices were normal. Extensive rheumatologic testing was also normal. Abdominal CT and abdominal MRI revealed an 8 cm left adrenal mass without signs of nearby tissues infiltration or lymph node involvement (Fig. 1), whereas cranial/thoracic CT and thoracic MRI showed no abnormalities. The functional imaging characteristics of the tumor was quite unspecific; in the abdominal CT, the adrenal mass had a density of HU~17 on the unenhanced phase and exhibited a slight gradual enhancement along with a nonspecific delay wash-out pattern both in the abdominal CT and MRI. The endocrine work up was normal: 24-hour urinary catecholamines (8 μg/24 h), metanephrines (258 μg/24 h) and VMA (3.0 μg/) were within normal limits. The 24-hour urinary cortisol level (1.1 mg/dL) and plasma aldosterone to renin ratio (AAR = 1.90) ruled out the possibility of hypercortisolism or primary hyperaldosteronism. The presence of a huge, 8 cm left adrenal mass with inconclusive imaging characteristics and hormonally inactive character deemed the tissue diagnosis mandatory, so a left laparoscopic adrenalectomy was scheduled. The operation was uneventful and the postoperative convalescence was excellent. The patient was discharged in excellent general condition on the third postoperative day.
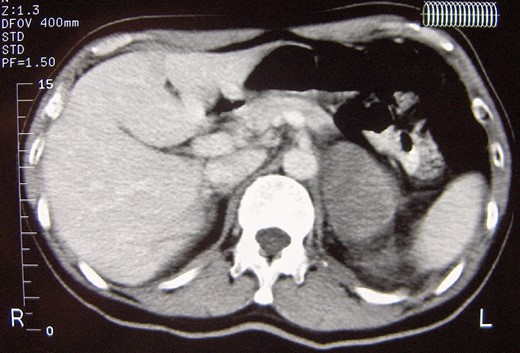
Abdominal computed tomography showing a well circumscribed 8 cm mass of the left adrenal gland with no signs of local spread or lymph node involvement.
Histologic sections
The surgical specimen consisted of a solid, soft, tan colored 10.5 × 7 × 4 cm tumor (Fig. 2). H&E sections showed a neoplasm confined to the adrenal gland consisting of tumor cells located inside dilated, thin-walled vascular spaces (Fig. 3). The neoplastic cells were large with irregular nuclei that exhibited atypia and had one or more nucleoli. Necroses were evident. Immunohistochemical stains showed that the neoplastic cells were positive for Vimentin, LCA, CD20 (Fig. 4), CD79a (Fig. 5) and negative for Cyrokeratins 7, 8, 18, 19, HMB45, S100, CD30, Myeloperoxidase, Inhibin, CD3, CD5, CD56, Chromogranin, CD31 and CD34. These findings were consisted with the diagnosis of primary adrenal intravascular large B-cell lymphoma. The patient was then referred to a Hematology Unit for further staging and treatment. Six cycles of R-CHOP were given lasting 21 days each: (Rituximab 375 mg/m2 iv (D1), Cyclophosphamide 750 mg/m2 iv (D1), Doxorubicin 50 mg/m2 iv (D1), Vincristine 1 mg/m2 iv (D1), P: Prednizolone 100 mg iv on D1-D5). After the completion of this chemotherapeutic regimen, a PET scan showed complete remission of the disease. Thereafter, the follow-up schedule consisted of CT studies every 6 months for 2 years and once annually, for the next 3 years. Despite the dismal prognosis, the patient 6 years after the initial diagnosis remains alive and in complete remission.
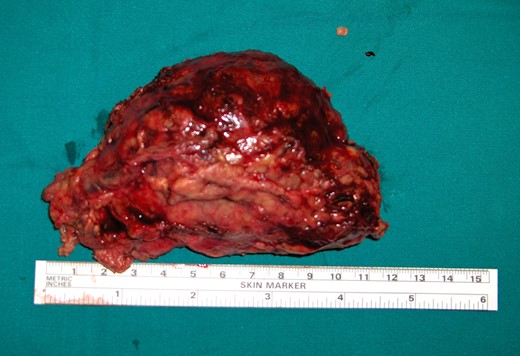
Macroscopic appearance of the left adrenal tumor.
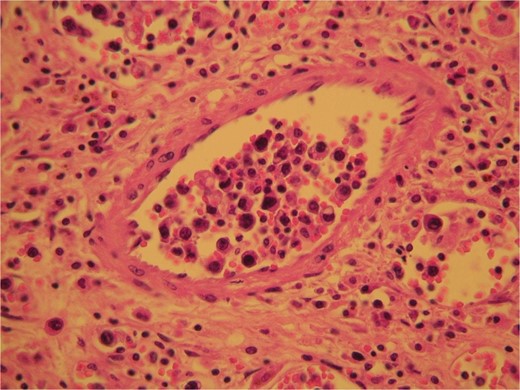
H&E sections revealed the presence of tumor cells inside dilated, thin-walled vascular spaces exhibiting minimal cytoplasm and irregular nuclei with atypia.
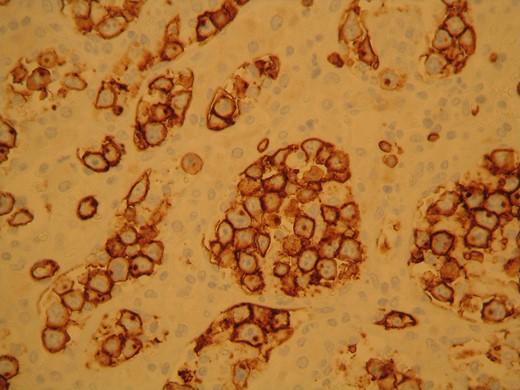
The neoplastic cells stained positive for CD20 confirming the presence of B lymphocytes.
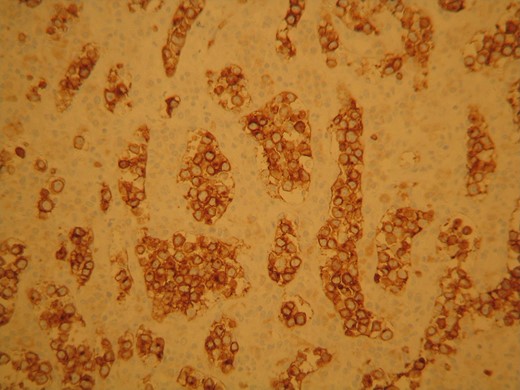
The neoplastic cells stained positive for CD79 characteristic for B lymphocytes presence.
DISCUSSION
IVLBCL is a rare type of large B-cell lymphoma characterized by selective growth of lymphoma cells within the lumen of vessels, particularly capillaries, sparing larger arteries and veins. The tumor occurs in adults, shows no sex predilection and carries a grave prognosis. It is usually widely disseminated in extranodal sites, however lymph nodes are usually spared. Clinically, it presents with two distinctive patterns: either a Western form with symptoms related to the affected organ or as an Asian variant with multiorgan failure, hepatosplenomegaly, pancytopenia and haemophagocytic syndrome. B symptoms are very common [2–4]. The mechanism for the intravascular growth pattern is still not known, however a lack of homing receptors and adhesion molecules including CD29 and CD54 has been hypothesized [5, 6]. Although many organs can be affected, brain and skin are especially susceptible. Adrenal involvement may be either isolated [7, 8] or bilateral leading to adrenal insufficiency [6, 9]. In our case, the adrenal tumor was confined to the left adrenal gland and was discovered incidentally in an abdominal CT during a work up for muscle weakness. The operative approach deemed necessary to obtain tissue diagnosis from this 8 cm hormonally inactive adrenal tumor that exhibited unspecific imaging characteristics. However, the choice of surgical access, whether open or minimally invasive, was puzzling due to the large size of the mass and the suspicion of malignancy. We opted for the laparoscopic approach, since the functional imaging characteristics were inconclusive and the radiologic findings showed neither infiltration of the surrounding structures by the tumor, nor any signs of lymph node involvement. Admittedly, this choice includes a very low threshold for conversion to open approach should any signs of infiltration, tumor capsule disruption or lymph node involvement are noted intraoperatively. Despite the documented dismal prognosis of this rare type of Non-Hodgkin Lymphoma, the patient remains alive and disease free, 6 years postoperatively.
As a conclusion, IVLBCL may be an unexpected diagnosis of an incidental adrenal mass. Laparoscopic adrenalectomy can be safely applied in cases with no signs of local or locoregional tumor extension. Despite its dismal prognosis, long-term survivors may exist as in our case.
Conflict of Interest statement
None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}