





Abstract
Osteopetrosis (OP) is hereditary X-linked, autosomal recessive (ARO), or autosomal dominant (ADO) skeletal disease. ARO has two subtypes, which are infantile malignant and intermediate type. ARO and X-linked OP have poor clinical outcome. ADO is called adult benign type because of the normal life expectancy, which has type I and type II. Here, the authors present an ADO patient with Chiari type I. Concomitant ADO with Chiari type I malformation is an extremely rare condition. Literature research yielded only one case report to date.
INTRODUCTION
Osteopetrosis (OP) has also called with its historical names as a Albers-Schönberg disease, which was firstly described by a German radiologist [1, 2].
The main cause of the disease is osteoclastic activity defect, which results hypersclerotic fragile bone [3, 4]. Autosomal recessive OP (ARO) has a two subtype, which are infantile malignant OP and intermediate type. Infantile malignant ARO has diagnosed within the first year of the life that has fatal clinical progression due to the bone marrow deficiency and infection. Intermediate ARO has rarely shown hematologic disorders. The life expectancy is poor [2, 3]. Bone changes occur more severely at the skull base. Important neurological symptoms are cranial verve palsies due to the obliteration of cranial nerve foramina such as vision loss, deafness, and facial paralysis [5]. The most common neurological finding is visual loss due to the optic nerve compression within the optic channel [6]. In contrast to the ARO, autosomal dominant OP (ADO) is described as adult benign type due to the normal life expectancy [2, 3, 7].
ADO diagnosis is given easily by recurrent bone fracture history, osteomyelitis, radiologic and laboratory findings, and genetic examination. The typical signs are bone within the bone on the end bones, sclerotic appearances on the long bone and multiple previous fracture lines. Other radiological findings are hypoplastic maxillary sinus, teeth deformity and multiple carious [8]. The genetic mutation in the chloride channel 7 (ClCN7) gene has been shown. ClCN7 gene mutations are heterogeneous, but the result is osteoclastic activity failure [4, 6].
CASE REPORT
A 25-year-old female patient presented with 1-year history of occipital headache. The headache was increasing with coughing, sneezing and bending over. She had also complaints of intermittent bilateral hand numbness and bilateral feet burning. She was evaluated with magnetic resonance imaging (MRI), which showed Chiari I malformation with a 7 mm descent of cerebellar tonsils (Fig. 1 a and b). Computed tomography demonstrated diffuse calvarial thickening and loss of the medullary space (Fig. 2). Her past medical history was consistent with left eye surgery due to amblyopia when she was a child and she was blind in her left eye. Radioactive 131I treatment had been given due to the Graves’ disease and hyperthyroidism; therefore, she was hypothyroidic and was using levothyroxine daily. Her mother also was operated on because of the Chiari malformation. She had four siblings, and they did not have any pertinent medical history. Her physical examination revealed prominent occipital area and midfacial hypoplasia. Increased cortical thickness in her long bones was also demonstrated (Fig. 3 a–c). There were no abnormal findings on neurological examination except of left eye amorozis. The patient underwent surgery for decompression of posterior fossa. Large enough posterior fossa craniectomy with C1 laminectomy was performed with SSEP and MEP monitoring. Dura was opened in ‘Y’ shape, and duraplasty was performed with the pericranium. Arachnoid was kept intact. Her headaches improved significantly in postoperative period. She developed superficial wound infection, which was treated with simple washout and antibiotics.
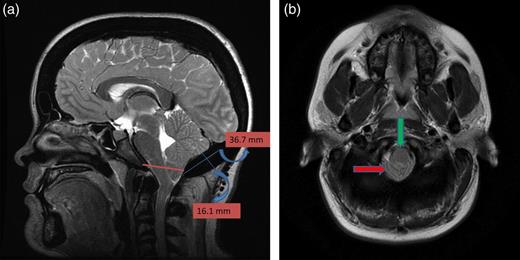
(a) Sagittal T2-weighted image showing cerebellar tonsillar herniation and diffuse thickening of the occipital bone. (b) Axial T1-weighted image showing compression on the upper cervical spinal cord (green arrow) and cerebellar tonsillar herniation (red arrow).
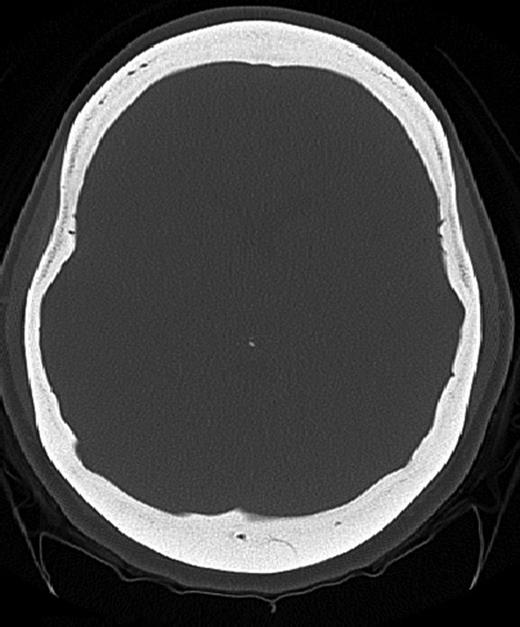
Axial CT image (bone window) showing diffuse calvarial thickening and loss of the medullary space.
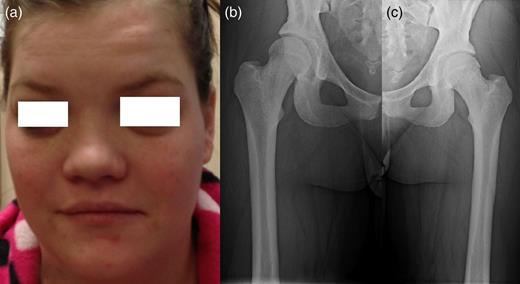
Patients' picture showing midfacial hypoplasia on the face (a), x-ray graphy of the right femur (b) and x-ray of left femur (c) showing diffuse cortical thickening.
DISCUSSION
OP is a rare and hereditary skeletal disease, which resulting from destructive or absence of osteoclastic activity [4]. Over all incidence is 1/250 000 births for ARO and 1/20 000 births for ADO [2].
This case is the second ADO case associated with Chiari type I, which presented with hindbrain headache and numbness on the upper extremity without other cranial nerve compression sign. MRI revealed brain stem compression, tonsillar herniation, occipital bone thickening and shallow posterior fossa. The first diagnosis was Chiari type I, but detailed history, endocrinologic and radiologic work-up yielded as an ADO, which was a final diagnosis.
Diagnosis is given with clinical and radiologic evaluation [8, 9]. Genetic examination confirms the clinical diagnosis if applicable. There are at least 10 genes, which has been identified. These diseases can be inherited as autosomal recessive, dominant or x-linked. ARO and x-linked OP have poor prognosis [2]. Balemans et al. [3] reported that untreated ARO patients died by the age of 4 years because of the pancytopenia and recurrent infection, which had a 75% rate.
ADO generally starts late childhood or adolescence [2–4]. There are two type describe, which have different clinical, biochemical and histological manifestation. Type I has osteosclerosis on the cranial vault, while Type II has end-plate thickening of vertebral body (Rugger Jersey spine) and end bones in the pelvis. Both the types are strictly family related and seen in late childhood [3, 10]. ADO patients are often asymptomatic. Symptoms are correlated with osteosclerosis and progressive with age. The fracture frequency is increased in type II and normal type I [1, 2, 4, 8, 10]. Cranial nerve compression is common in ADO type I [8].
De Oliveira et al. [4] reported that there is no need to perform genetic study to confirm the disease. Tohidi and Bagherpour [10] suggest that ADO is rare condition, which can be asymptomatic. Therefore, accurate diagnosis can be achieved with the proper clinical, endocrinologic and radiologic investigation. Calcium, phosphorus and alkaline phosphatase levels are usually within the normal limits in the benign OP [7, 10].
Dlouhy and Menezes [7] proposed in their report that even if their report is the first case ADO with Chiari, further study is needed. In their report, the cause of the Chiari malformation in the OP explained as unclear. Hypotheses include mass effect from the significant calvarial thickening and subsequent downward tonsillar herniation. Further, occipital bone thickening may lead to a smaller posterior fossa. Moreover, the absence of the osteoclastic activity leads to underdeveloped occipital bone, and this results in a smaller posterior fossa [7].
In conclusion, ADO with Chiari type I malformation is a rare condition. However, patients can present with only Chiari symptoms without other cranial nerves compression symptoms; therefore, ADO can be overlooked for differential diagnosis. Symptomatic Chiari malformation needs surgical intervention, but it should be kept in mind that the patients with Chiari malformation should investigate differential diagnosis for ADO. Whole skeletal survey must be done, whether there are sclerotic chances, thickening skull base, bone within the bone appearance, Rugger Jersey spine, Erlenmeyer flask deformity on the long bone and fracture line on the bone.
CONFLICT OF INTEREST STATEMENT
None declared.

{kind=link}
{kind=link}
{kind=link}