





Abstract
The vast majority of adult primary cardiac tumours (75%) are benign. The differentiation between malignant and benign lesions based on imaging is often difficult. Furthermore, it is challenging to distinguish between a cardiac myxoma and a myxosarcoma histologically. We report the case of a 48-year-old female who underwent resection of myxoma. Fourteen months postoperatively, she developed dyspnoea and evidence of local recurrence was observed. An open biopsy was performed and compared with the initially resected specimen. A primary cardiac myxosarcoma was diagnosed. Extended resection of the tumour including a part of the left atrium and the left lung was performed. Follow-up at 4 years shows no radiological evidence of any further recurrence and the patient is satisfied with a good quality of life. Despite the infrequent nature and particularly in view of the poor prognosis of cardiac myxosarcoma with a median overall survival of ∼12–17 months, we were able to demonstrate in our case that, with an extensive medical and surgical therapy and an interdisciplinary approach, a long-term disease-free survival can be achieved.
INTRODUCTION
Primary cardiac neoplasms are rare [1]. Metastases from other primaries or direct invasion of the heart are more common. The vast majority of adult primary cardiac tumours (75%) are benign, but the proportions depend on the location of the mass [2]. Half of the primary tumours of the right atrium are malignant, while a majority of left atrium tumours are benign and mostly consist of myxomas (50–75%) [3]. The differentiation between malignant from benign lesions based on imaging is difficult. It is equally challenging to distinguish between a cardiac myxoma and a myxosarcoma histologically. Myxosarcoma may be misdiagnosed as myxoma until the neoplasm recurs due to gross and histological similarities between them [4]. Despite the infrequent nature and particularly in view of the poor prognosis of malignant disease with a median overall survival of ∼17 months, an accurate preoperative assessment, prompt diagnosis and aggressive treatment are mandatory.
We report the first case of primary cardiac myxosarcoma associated with long-term survival. The tumour was initially diagnosed as a benign myxoma. However, a review of the initial histology and the recurrence of the neoplasm after primary resection established the diagnosis of a primary cardiac myxosarcoma.
CASE REPORT
A 48-year-old female presented with progressive dyspnoea, non-productive cough, weakness, dizziness and loss of weight stretching over a period of 4 weeks. The past history was non-specific. Echocardiography revealed a large, intracavitary, broad-based mass attached to the left interatrial wall and the roof of the atrium with extension to the superior pulmonary vein. A left atrial myxoma was suspected. After ruling out an associated coronary heart disease, the patient underwent resection of the atrial septum including the atrial roof with subsequent patch reconstruction of the left atrium and the left upper pulmonary vein. Histologically, the tumour had a myxoid background with spindle-shaped cells, moderate cellularity and atypia (Fig. 1A). There were no definitive areas of vascular patterns. Based on these histological findings and the preoperative imaging, a diagnosis of myxoma was presumed and subsequently no further immunohistological staining was carried out. No further adjuvant therapy was performed. Regular follow-up in an interdisciplinary team included clinical evaluation, chest-CT and echocardiography at a 6-month interval. At 14 months, evidence of local recurrence was observed. An open biopsy was performed and compared with the initially resected specimen (Fig. 1B). The diagnosis of myxosarcoma was confirmed. Following four cycles of neoadjuvant chemotherapy with iphosphamide and doxorubicin, the patient underwent a complete en bloc resection of the tumour mass including a left pneumonectomy due to massive infiltration of the hilus (Fig. 2) through a left hemi-clamshell incision under cardiopulmonary bypass. The excised polyobulated mass revealed histologically, high mitotic activities, up to 8 per high power field. By immunohistochemistry, the tumour cells were diffuse positive for vimentin, moderately positive for calponin, smooth muscle antigen and desmin. Recovery was uneventful. Subsequent follow-up at 4 years shows no radiological evidence of any further recurrence. The patient is satisfied with excellent quality of life.
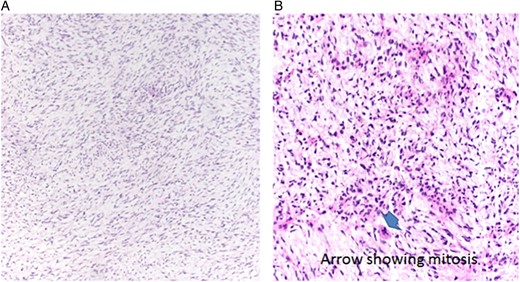
(A) Histological image of myxoma showing a cellular background with multiple spindle-shaped cells and bland nuclei with myxoid matrix. (B) Histological image of myxosarcoma showing hyperchromatic cells, atypia and mitosis (arrow), ×20, H&E
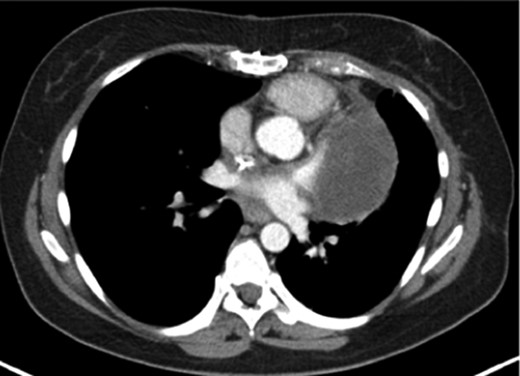
CT showing the tumour with infiltration of the main trunk of the left pulmonary artery with involvement of the hilus
DISCUSSION
Patients with primary cardiac sarcoma present with identical signs and symptoms of a left atrial myxoma. Echocardiography has been shown to be crucial in the diagnosis of endocardial lesions. Although non- neoplastic intracardiac lesions like vegetation and thrombi are not difficult to distinguish from benign atrial myxomas, it may be nearly impossible to definitively distinguish between atrial myxoma and myxosarcoma. In evaluating a mass in the left atrium, a non-septal origin of the mass, extension into the pulmonary vein, multiple masses, broad attachment on the left atrial wall and semisolid consistency should raise the suspicion of a malignant nature of the neoplasm [5]. A sarcoma with significant myxoid changes can histologically mimic a myxoma, which occurred in our case. Myxoma and myxosacarcoma are both characterized by extracellular deposition of proteoglycans and usually located in the left atrium, which adds up to the difficulty in distinguishing between them. The most important histological criterion to establish the diagnosis of myxosarcoma is the absence of the typical cords, rings and capillary structures formed by myxoma cells. The postulated theory about a malignant transformation of myxoma is quite controversial. The postulated reasons to doubt this transformation are, first, composite tumours consisting of sarcoma and myxoma probably do not exist. Secondly, a myxoid background may occur in all types of cardiac sarcomas and may be a consequence of intracavitary location rather than a reflection of histogenesis [4].
Cardiac malignancies are difficult to treat with any modalities (operation, chemotherapy, radiation or transplantation) due to the high metastatic potential and unresectability at presentation. Therapy usually consists of an induction treatment followed by complete resection if possible. Despite the use of an aggressive therapy concept with chemotherapy and radiation, isolated or combined with surgery, advantages in terms of quality or quantity of life have not been clearly demonstrated. Indeed, the long-term survival of patients with primary myxosarcoma of the heart remains poor. Factors associated with an increased survival are left-side location, a mitotic rate of <10 mitoses per high-powered field and no necrosis, whereas tumour histotype does not seem to affect the prognosis [4].
Previous reports on patients undergoing surgery for cardiac myxosarcomas have shown that the initial operation is commonly successful, but few patients survive >12–17 months [5]. A high degree of suspicion is required to arrive at a preoperative diagnosis of cardiac myxosarcoma. In our case, we were able to demonstrate that, with an extensive medical and surgical therapy and an interdisciplinary approach, a long-term disease-free survival can be achieved. It is to be emphasized that histological features of primary cardiac myxosarcoma are quite distinctive in comparison with myxoma, and further research is required to define the accurate histogenesis of cardiac myxosarcoma.
CONFLICT OF INTEREST STATEMENT
None declared.

{kind=link}
{kind=link}