





Abstract
Neonatal cushing syndrome (NCS) is a rare disease that results from prolonged exposure to high cortisol levels. McCune–Albright syndrome (MAS) is an exceedingly rare genetic disorder characterized by cafe-au-lait skin spots, bone fibrous dysplasia and multiple endocrinopathies. We describe a case of a premature neonate with Intrauterine Growth Retardation who presented with hypercortisolemia, neonatal transaminitis and cardiac dysfunction. Further evaluation revealed significant bilateral adrenal hyperplasia leading to the diagnosis of NCS as part of MAS. Despite maximum medical therapy, including metyrapone, the baby’s refractory hypertension, hyperglycemia and persistent failure to thrive (weight of 1.4 kg at corrected age 38 weeks) necessitated bilateral adrenalectomy. This case did not initially demonstrate the classic MAS triad, notably, the absence of skeletal manifestations. There has been no previous description of a baby who has had all the early life-threatening features present and survived beyond 18 months. This case highlights the severity of the phenotype and the challenges involved in diagnosing and treating NCS and MAS in neonates.
INTRODUCTION
Neonatal cushing syndrome (NCS) is a rare disease defined by the presence of multi-system signs and symptoms that develop as a result of prolonged exposure to high cortisol levels [1]. The most common manifestations are decelerated growth and facial roundness. The Endocrine Society recommends the use of tests with high diagnostic accuracy to confirm NCS; namely two of the following four tests: urine cortisol, late-night salivary cortisol, overnight or 48-h dexamethasone suppression test. Multiple Adrenocortical Diseases (ACDs) might cause NCS including McCune–Albright Syndrome (MAS) [1, 2]. MAS is an exceedingly rare genetic disease characterized by the triad of Café-Au-Lait skin spots (CAL), bone Fibrous Dysplasia (FD), multiple endocrinopathies and rarely hepatobiliary dysfunction and cardiac diseases. When NCS is associated with MAS, and given the probability of spontaneous resolution of hypercortisolemia, medical management is preferred to surgery, with the benefit of preserving adrenal functions [3, 4]. Here is a description of a premature baby girl with a history of Intrauterine Growth Retardation (IUGR), who had hypercortisolemia, neonatal transaminitis and cardiac dysfunction. Subsequently, the baby was found to have significant bilateral adrenal hyperplasia leading to NCS, precocious puberty and café-au-lait skin spots, all as part of MAS.
CASE PRESENTATION
A female neonate was born at 31 + 5 weeks of gestation by elective cesarean section due to severe symmetrical IUGR. Baby weighed 1.2 kg at birth. APGAR scores were 7 and 8, at 1 and 5 min, respectively. She was the fourth child of consanguineous parents who had a previous IUGR baby. Baby was admitted to Neonatal Intensive Care due to respiratory distress and required breathing support, Continuous Positive Airway Pressure for 5 days. At 3 days of age, she was noted to have high Blood Glucose (BG) (10 mmol\L). On day 7, her liver function tests were noted to be deranged with progressively elevated alanine transaminase (ALT) (peak 950 U/L), gamma glutamate transaminase (840 U\L) and bilirubin (total: 11.1 and direct: 10.5 umol\L). Baby’s urea levels were also raised (12, Ref: 2–8 mmol\L). Baby had a standard workup for sepsis, Toxoplasmosis, Rubella, Cytomegalovirus and Herpes infections (ToRCH), Epstein-Barr virus (EBV), parvovirus and viral hepatitis, all of which were negative. Her thyroid and bleeding disorders screens were also negative. Metabolic screen including neonatal spot test, was normal. An abdominal Ultrasound scan was unremarkable.
Between day 21 and 35, systolic blood pressure (BP) was noted to be consistently elevated ≥100 mmHg (>95th centile for her age). Renal artery doppler ultrasound and urine catecholamines were normal. A cardiac echocardiogram detected left ventricular hypertrophy. She was started on Amlodipine 400 mic/kg/twice a day. Subsequently, her BG spiked to 19.1, therefore amlodipine was discontinued and insulin drip were initiated. The baby was also noted to have full cheeks; double chin, resistant hypertension and poor weight gain (only 200 g since birth). Further laboratory investigations showed a high level of random cortisol 1839 nmol, (normal: am 133–537, pm 68–327), low Adrenocorticotropic hormone (ACTH) and increased testosterone.
NCS was confirmed by elevated midnight cortisol level, dexamethasone suppression test and bilaterally enlarged adrenals on contrast-enhanced computed tomography (CT) scan as shown in Fig. 1. For NCS Metyrapone was introduced with a gradually increasing dose to 60 mg four times a day (QID). Though initially some symptomatic relief was accomplished, as a side effect baby developed a progressive increase in ALT to 1400. However, there was no long-lasting significant improvement in the Cortisol level. Cortisol remained very elevated at 2400 nmol as illustrated in Fig. 2, along with refractory hypertension, hyperglycemia and persistent failure to thrive (FTT) (weight 1.4 kg, length 36.5 cm at 7 weeks of age). Ketoconazole was not considered for treatment because of the elevated ALT.
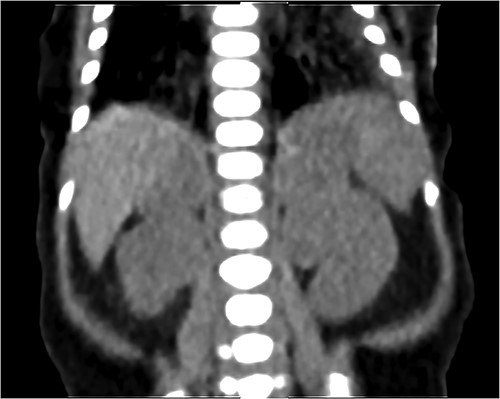
Coronal CT scan with IV contrast shows a uniform lobular enlargement of both adrenals (red arrows).
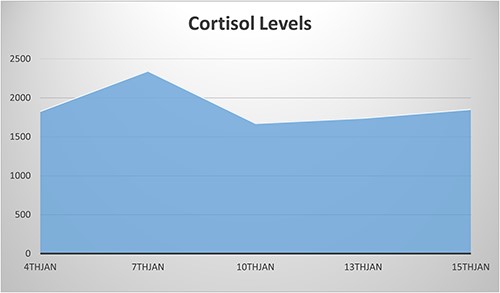
Persistent elevated cortisol level despite using Metyrapone until the date of the surgery (10th Jan).
Therefore, surgical management in the form of bilateral adrenalectomy was undertaken. The left adrenal gland measured 3 × 4 cm, and the right 3 × 2 cm with a combined weight of 4.59 g. The normal combined weight of the adrenals in an average-sized full-term neonate is 2–3 g [5]. She had an uneventful surgical post-operative course. Histopathology of the adrenal glands confirmed multi-micronodular ACD. McCune–Albright syndrome was suspected but genetic testing, including whole exome sequencing and BRCA1 were inconclusive.
Subsequent to the surgery ALT and cortisol both decreased over time, but her BP remained moderately high, albeit less volatile. Therefore, further cardiac evaluation showed the baby to have coarctation of the abdominal aorta and was therefore started on Propranolol.
Following the surgery, she gained weight but remained small for age. She was discharged home 1 month after the surgery with Propranolol and steroid replacement; hydrocortisone and fludrocortisone.
On follow-up visits, hyperpigmented spots with irregular margins were noted on baby’s back at 4 months of age (Fig. 3), vaginal blood spotting was reported at 1 year of age and was found to have global developmental delay. She also had markedly persistent elevated ALT as well as raised bile acids. Hepatobiliary ultrasound and CT were unremarkable. Consequently, the baby was started on ursodeoxycholic acid. She is currently considered to be a case of MAS and continues to be followed up in our outpatient department.
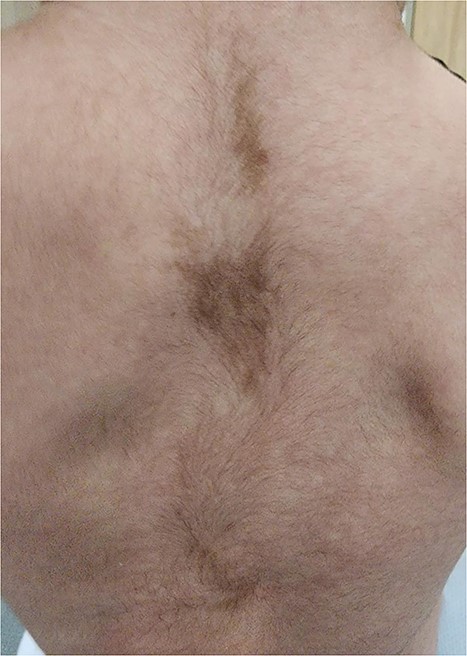
CAL spots; macules with jagged irregular edges (white arrows).
DISCUSSION
NCS is the constellation of signs and symptoms that result from the chronic effects of hypercortisolemia in the neonatal period. The initial presentations in children are decelerated growth or FTT, which is a feature of many common neonatal diseases. More specific signs include buffalo hump, moon face, facial plethora, hirsutism, decreased bone mineral density and impaired glucose tolerance [1–3]. Hypertension can be a significant finding as it can be resistant to medical management and may even lead to death due to cerebral vascular accidents [6, 7]. Many ACDs may cause hypercortisolemia, these include Li-Fraumeni Syndrome, Carney Complex, Beckwith-Wiedemann Syndrome, Multiple Endocrine Neoplasia type 1, and Familial Adenomatous Polyposis Syndrome. Nevertheless, the majority of these illnesses are not known to cause NCS [1, 2].
NCS due to MAS has been reported in the literature and has various outcomes, ranging from spontaneous resolution to the need for adrenalectomy or unfortunate event of death [8–10]. Increased cortisol levels plausibly may induce suppression of the immune system, thus increasing the risk of fatal opportunistic infections, particularly Pneumocystis Carinii. Prophylactic antibiotics can be beneficial in such cases [1, 11].
GNAS1 is a gene that encodes Gαs protein, which in turn regulates the activity of Cyclic AMP (cAMP). cAMP controls the production of numerous hormones that markedly influences various cells. GNAS1 somatic gene mutations, that cause MAS, lead to over-manufacture of hormones by sempiternal-activated cAMP, manifesting in multi-organ dysfunction [1, 3]. Due to the mosaicism of the disease’s genetic etiology, a wide spectrum of skeletal and extraskeletal manifestations are demonstrated, and the negative result of the gene study does not eliminate the likelihood of MAS. Consequently, diagnosis is generally established clinically [12].
Neonatal exhibition of MAS is very rare, and the typical initial presentation in pediatrics is precocious puberty or FD [3]. In general, liver dysfunction and cardiac manifestations are exceedingly rare, and NCS is the rarest among other endocrinopathies, which is a leading pitfall in reaching the diagnoses and the management [3, 4, 8].
CAL patches often appear near or at birth. As such, they can be early evidence of MAS [8, 9, 18]. Although Neurofibromatosis1 (NF1) and MAS both show CAL spots, NF has fewer and larger macules and smooth borders. MAS has smaller, more numerous macules with jagged irregular edges [3, 11].
To date, there have been only two reported cases of total bilateral adrenalectomy in preterm infants for the treatment of NCS. One of these is an infant with a weight of 1.6 kg at the time of surgery [13], while the other with a weight of 2.13 kg [14]. Both cases did not have MAS. Therefore, our case represents the smallest surviving reported preterm infant with MAS to have undergone neonatal adrenalectomy at 1.4 kg.
Recent reports have indicated that hepatobiliary dysfunction and cardiomyopathies are important risk factors for mortality and morbidity in MAS. Only a few cases with cholestasis secondary to MAS during infancy or early childhood have been reported. Consequences are unpredictable, ranging from temporarily elevated transaminases to liver transplant [12, 15–17]. Two cases with ventricular hypertrophy, as a part of MAS syndrome, eventually ended up being fatal [8, 9], and two survived [18, 19]. The causes of death were due to pulmonary embolism and severe respiratory infection.
Our case did not initially demonstrate the classic MAS triad: bone dysplasia, CAL patches or precocious puberty. She presented with rare manifestations; cardiac, liver and adrenal symptoms. This is particularly noteworthy as according to two reviews by Corsi A et al. [9] and Brown RJ et al. [10], there are no previously documented cases of individuals alive beyond 18 months of age although having all the early life-threatening features that were present in this patient during the neonatal period. These include IUGR, prematurity, bilateral adrenalectomy in a very low weight due to NCS, transaminitis and ventricular hypertrophy. This case highlights the severity of the phenotype and the challenges involved in diagnosing and treating MAS in neonates.
CONCLUSION
This case report represents the only surviving case of NCS and MAS with multiple severe phenotypic features. Numerous potential pitfalls in diagnosis and management are highlighted and appropriate management is discussed.
CONFLICT OF INTEREST STATEMENT
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
FUNDING
This paper did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
References
Author notes
Muhammad Eyad Ba’ath, senior author

{kind=link}
{kind=link}
{kind=link}