





Abstract
The Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome is a congenital disorder caused by the abnormal absence of paramesonephric ducts. The main characteristics of this syndrome include hypoplasia or aplasia of the uterus, absence of the cervix and upper part of vagina. Otherwise, ovaries and fallopian tubes have normal functions. A 9-year-old girl presented to the emergency department by acute abdominal pain. Based on a high probable diagnosis of ovarian torsion, surgical exploration was accomplished demonstrating rudimentary or aplastic uterus, hypoplasia of the left adnexa and torsion of the right ovary. In consideration of MRKH syndrome, further assessments were done and the diagnosis was confirmed.
As this syndrome is rare and there is a probability of ovarian torsion caused by malformation of the ovarian ligament, physicians should be aware of this syndrome to diagnose it earlier and preserve the ovarian tissue.
INTRODUCTION
The Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome is a congenital disorder with an incidence rate of 1 in 4500 female live births [1]. The etiology of MRKH syndrome is the abnormal absence of paramesonephric (Mullerian) ducts during embryogenesis [2], which results in main characteristics of this syndrome, including congenital hypoplasia or aplasia of the uterus, absence of the cervix and two-thirds upper part of the vagina. Otherwise, ovaries and fallopian tubes have normal functions with normal female karyotype of 46 XX [3]. This syndrome typically is diagnosed at puberty presenting by primary amenorrhea with normal pubarche and thelarche [4].
Two subtypes of this syndrome have been described: patients of type 1 demonstrate the only malformation of genital parts, but there is a MURCS (Müllerian, Renal, Cervicothoracic Somite) association abnormality in patients of type 2 [5].
In addition, a VCUAM (Vagina, Cervix, Uterus, Adnexa—associated Malformation) classification has been presented in 2005 to classify patients based on genital and related malformations [6].
Although both ovaries are present and have normal function in this syndrome, rarely ovarian anomalies have been reported by the prevalence of 5–10% including hypoplasia, streak ovaries, malposition and tumors [4].
Our goal is to describe of a rare case of MRKH syndrome in prepuberty age presenting with right ovarian torsion and hypoplasia of the left adnexa, which meets the criteria of group A2a of VCUAM classification. The main characteristic of this subgroup is unilateral hypoplasia or gonadal streak with a rare prevalence of 3.5% in all cases of MRKH syndrome [6].
CASE REPORT
A 9-year-old girl presented to the emergency department complaining of nausea, vomiting, and abdominal pain for about 1 day before admission. No fever, chill and urinary symptoms were reported.
Her past medical history was unremarkable and she did not have any history of menstruation.
On examination, tenderness of lower quadrants was detected. Since the hymen was intact, vaginal examination was not performed but a rectal examination revealed tenderness of the right adnexa without any blood in the stool.
On laboratory data, the WBC count was 11500/mm3. Other exams were unremarkable. To rule-out the diagnosis of appendicitis, ultrasonography was done that revealed torsion of the left ovary with minimal free fluid in the pelvis. Importantly, the radiologist could not observe the uterus and right ovary. Other organs such as the liver, gall-bladder, both kidneys and bladder were reported normal (Fig 1).
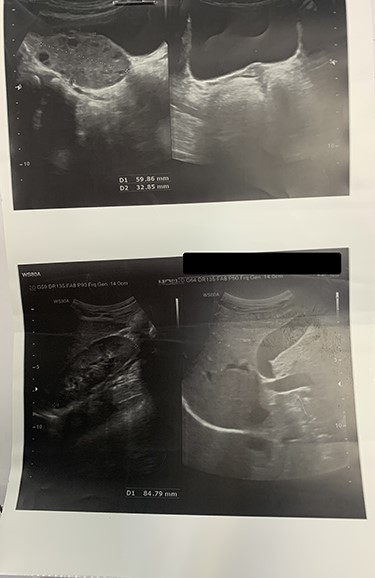
Ultrasonography shows the enlargement of the right ovary in size of 59 × 32 mm with increased stroma vulnerable of ovarian torsion. Uterus and the left ovary cannot be seen.
Based on a high probable diagnosis of ovarian torsion, surgical exploration was accomplished demonstrating bilaterally rudimentary or aplastic uterus, unilateral hypoplasia of the left adnexa, and torsion of the right ovary containing a simple cyst. Since the right utero-ovarian ligament was longer than normal size, the right adnexa was twisted and dislocated in the left lower quadrant, so the radiologist misreported torsion of the left adnexa (instead of the right side) (Fig. 2).
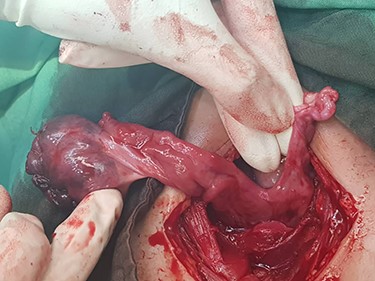
Findings of laparotomy: Torsion of right ovary and hypoplasia of the left adnexa. Uterus cannot be seen. Note the long ligament of the right ovary.
Detorsion and cystectomy of the right ovary were done and plication of the utero-ovarian ligament was performed to prevent recurrent torsion.
On the follow-up, magnetic resonance imaging revealed normal anatomy of spines and kidneys and absence of the uterus. The result of pathology reported a simple cyst of the right ovary. Also, karyotype of the patient was normal and reported as 46 XX.
DISCUSSION
As the prevalence of MRKH syndrome is low, most gynecologists are not familiar with this syndrome. Our goal is to introduce this rare case and its presentation.
Typically, diagnosis of MRKH syndrome is at puberty following assessment of primary amenorrhea in patients who manifest secondary sexual characteristics. [7] The main etiology of MRKH syndrome is the abnormal development of paramesonephric ducts [8], as the ovaries originate from genital ridges, we expect to have a normal gonadal function in these cases. [9]
Our case is a rare manifestation of MRKH syndrome with right ovarian torsion and left ovarian hypoplasia, which belongs to group A2a of VCUAM classification with a prevalence of 3.5% among all subtypes. [6]
In recent studies, some cases of MRKH syndrome who presented with torsion of adnexal mass and rudimentary uterus with Leiomyoma were reported [10–12]. In one literature, a patient with MRKH syndrome presented with one-sided adnexal torsion and ectopy of the other side [8], but our patient is a rare case of MRKH syndrome with the diagnosis of the right ovarian torsion and concomitant left side ovarian hypoplasia.
As mentioned before, this syndrome is rare and the probability of ovarian torsion is mostly due to malformation of the ovarian ligament. On the other hand, appendicitis and ovarian torsion have similar presentations, so the physicians should be aware of this rare syndrome so that they can diagnose and preserve the ovarian tissue by emergent surgical intervention since these patients could have the chance of pregnancy by induction of ovulation and surrogacy in future.

{kind=link}
{kind=link}