





Abstract
Adrenal sarcomatoid carcinoma (ASC) is a very rare aggressive variant of adrenocortical carcinoma showing carcinomatous and sarcomatous differentiation. It is a poorly differentiated carcinoma with poor prognosis. The diagnosis requires careful histological and immunohistochemical investigation. We describe a new case of ASC to raise awareness on this extremely rare entity. A 27-year-old woman presented with a right flank pain. Imaging revealed a tissular mass of the right adrenal gland without metastases. After adrenalectomy, histology revealed sheets of epithelioid cells that stained for synaptophysin and Melan-A; and spindled cells staining for S-100. We have reported the clinical and histopathological features of ACS’s case; as it is an extremely rare cancer with a challenging diagnosis. There is a need for a further understanding of ASC’s biology to improve it poor prognosis.
INTRODUCTION
Adrenal sarcomatoid carcinomas (ASC) are rare aggressive malignant neoplasms of the adrenal gland, showing both carcinomatous and mesenchymal differentiation [1]. It has a poor prognosis as most of patients have distant metastasis at the time of diagnosis [2]. The first case of ASC was reported in 1989 by Collina et al. [3]. To the best of our knowledge, only 20 cases have been reported in the English literature to date [1]. Herein, we describe a new case of an ASC and a review of the literature to raise awareness on this extremely rare entity which presents with a diagnostic challenge.
CASE PRESENTATION
A 27-year-old woman presented with a 6 months history of a right flank pain and a moderate weight loss. She had no remarkable medical history. Upon physical examination, she had stable vital signs, without adrenal hormonal hypersecretion’s signs. The patient’s blood cell counts, serum electrolyte levels, renal and liver functions, 24-h urinary catecholamines, cortisol, testosterone, dihydroxyepiandrosterone sulfate and aldosterone levels were all within normal limits. Abdominal computed tomography (CT) scan showed a 12.5 × 9 × 7.5 cm tissular heterogeneous mass, with irregular borders, of the right adrenal gland, with calcifications (Fig. 1). The routine staging was negative for metastases.
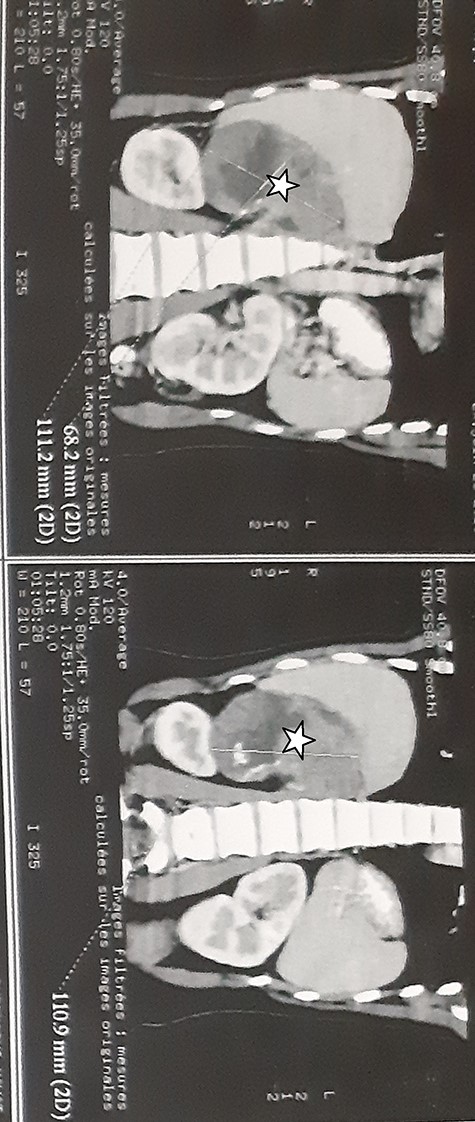
Abdominal CT scan showed a 12.5 × 9 × 7.5 cm tissular heterogeneous mass, with irregular borders, located in the right adrenal gland (white star).
She underwent an adrenalectomy. On gross, the specimen weighed 660 g and measured 12 × 9 × 7 cm. It was well circumscribed, firm, beige-colored, with extensive hemorrhage and necrosis at sectioning; completely effacing the adrenal gland. Hematoxylin–eosin stained sections revealed diffuse sheets of epithelioid and pleomorphic/spindled cells. The epithelioid component consisted of sheets and nests of polygonal cells with clear and eosinophilic (40%) cytoplasm resembling adrenocortical cells with loss of lobulation (Fig. 2A). There was marked nuclear atypia (Nuclear grade III) with high mitotic activity (5/10 high-power fields [HPF]). The pleomorphic component (40% of tumor) consisted of fascicles of spindled and ovoid cells. Nuclei were highly pleomorphic with dense heterogenous chromatin (Fig. 2B). Some multinucleated neoplastic giant cells were identified. The mitotic count was higher (12 mitoses/10 HPF). The Weiss score was 6. On immunohistochemistry, epitheliod cells showed strong, diffuse positivity with Melan-A and synaptophysin. Spindled cells were focally strongly positive for synaptophysin, Melan-A and PS100 (Fig. 3). Immunostains for AE1/AE3, HMB-45, inhibin, desmin, vimentin, caldesmon and SMA were negative in both components. Unfortunately, genetic analysis was not performed, because it is not available in our institution. After a follow-up of 6 months, there was no evidence of local recurrence on CT.
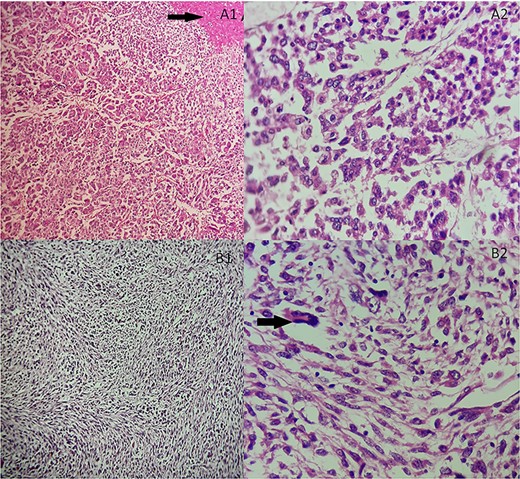
(A) Adrenocortical sarcomatoid carcinoma histological features Carcinomatous component showing sheets and nests of epithelioid cells and necrosis (black arrow). (A1) Hematoxylin and eosin, ×100. (A2) Hematoxylin and eosin, ×400. (B) Adrenocortical sarcomatoid carcinoma histological features: sarcomatous component consisted of spindle-shaped and ovoid cells arranged in fascicular pattern. Associated to some multinucleated neoplastic giant cells (black arrow). (B1) Hematoxylin and eosin, ×200. (B2) Hematoxylin and eosin, ×400.
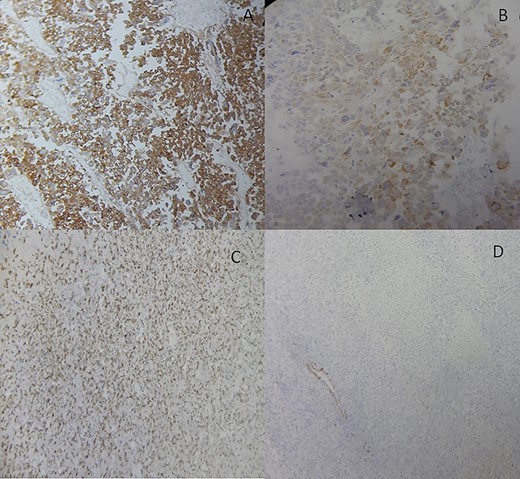
Adrenocortical carcinosarcoma immunohistochemistry: The carcinomatous areas are synaptophysin positive (A); Melan-A positive (B); PS100 positive (C) and SMA negative (D) (Immunohistochemical stain, ×200).
DISCUSSION
ASC is a very rare malignant tumor [4] (only 20 cases reported in the literature to date) [1]. ASC represents the least common variant of ACC [5]. Primary ASC mainly occurs in middle-aged patients (23–79 years) with a sex ratio (male:female) of 1:1. [6]. Our patient was a middle-aged woman. As in the present case, these carcinomas are usually non-functioning tumors [5] with non-specific symptoms including: abdominal masses; abdominal pain, weight loss and asthenia. When functional, following symptoms may occur including: severe hypertension; Cushing’s syndrome; feminization or masculinization [6]. ASC pre-operative diagnosis remains challenging [7]. Adrenal biopsy is not routinely carried out for suspicious adrenal masses, the accurate diagnosis is made after surgical exploration [8], by histological and immunuhistochemical study [7]. Sarcomatoid carcinoma are composed of malignant epithelial cells and specialized mesenchymal cells. Some cases show osteosarcomatous, rhabdomyosarcomatous or chondrosarcomatous differentiation [4]. The Weiss score is the most widely used system. It comprises nine parameters: (1) nuclear grade (Fuhrman III or IV), (2) mitotic rate of >5/50 HPFs, (3) abnormal mitoses, three refer to tumor structure (4): ≥25% clear cells, (5) >1/3 diffuse architecture, (6) necrosis; (7) venous invasion, (8) sinusoidal invasion and (9) capsular invasion. The epithelioid component shows features of conventional ACC and meets four or more of the Weiss histological criteria [2]. In our case, the WEISS score of the epithelioid component was nine and the sarcomatoid component did not show any recognizable differentiation. Definitive diagnosis requires immunostaining. Several markers are of diagnostic value including: Melan-A, synaptophysin, calretinin; as they confirm the adrenal phenotype of the tumor [7]. Epitheliod cells are also immunoreactive for vimentin, inhibin, neuron-specific enolase and occasionally positive for AE1/AE3. The laters are negative for chromogranin and EMA. According to previous studies, sarcomatous component may be positive for desmin, myogenin, HHF35, vimentin, myoglobin, caldesmon, smooth muscle actin, AE1/AE3, EMA, HMB-45, PS 100, inhibin and chromogranin [7, 8]. In the present case, spindled cells were focally positive for synaptophysin and Melan-A. These findings are similar to those reported by Sasaki et al. [7], which supports that sarcomatous component originated from the adrenocortical carcinoma [7].
The histological diagnosis of ASC, especially when mononophasic, is challenging. In fact, they can be indistinguishable from adrenal sarcomas and retroperitoneal sarcomas with adrenal extension (e.g. liposarcoma, rhabdomyosarcoma etc) [9]. The differential diagnosis also includes extra-adrenal carcinosarcomas and metastases with sarcomatoid elements. Thus, the diagnosis is based on a careful histological and immunohistochemical analysis and in some cases, on genetic analysis [7]. Adrenocortical carcinomas show variant genetic aberrations, including gains in chromosomes 4, 5, 7, 12, 16, 19 and 20, and losses involving 1p, 2q, 11q, chromosome 17, 13 and 22 [4]. ASCs are aggressive tumors with a very poor prognosis. Frequent metastases have been reported, post-operative survival ranges from 3 to 12 months after surgical treatment in the majority of patients. The longest postoperative survival (17 months) with no evidence of metastasis was reported by Mark et al. [10]. Radical surgical excision is the only curative treatment. Adjuvant chemotherapy is suggested after the first surgery, when distant metastasis or local invasions are absent [8]. To date, no evidence-based consensus have been reported regarding best oncological treatment of ASCs. Epithelial to Mesenchymal Transition (EMT)-related markers and stem cell factors might be novel therapeutic targets [5].
In conclusion, we have reported the clinical and histopathological features of this extremely rare cancer. Up to present, no standard efficient treatment regimens have been clearly identified. There is a need for a further understanding of ASC’s biology to improve its poor prognosis.
ACKNOWLEDGEMENTS
Not applicable.
COMPETING INTERESTS
The authors declare that they have no competing interests.
FUNDING
No funding source was needed.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
CONSENT
Informed consent was obtained from the patient. Guarantor is the corresponding author.
AVAILABILITY OF DATA AND MATERIALS
Not applicable.
AUTHORS’ CONTRIBUTIONS
All authors read and approved the final manuscript.

{kind=link}
{kind=link}
{kind=link}