





Abstract
This case describes a 9-year-old male who presented to general surgical clinic with a 3-year history of persistent natal cleft swelling, previously unsuccessfully treated as a pilonidal abscess in the community with multiple courses of antibiotics. In clinic, a 50 × 30-mm soft tissue swelling was found in the natal cleft and a clinical diagnosis of a pilonidal cyst was made. A cream-coloured solid mass measuring 50 × 35 × 30 mm was subsequently excised under general anaesthetic, with specialist histology and immunostaining confirming an unexpected diagnosis of a subcutaneous extraspinal myxopapillary ependymoma, a tumour usually found in the neuraxis. Given the atypical anatomical site of the tumour, the case presented a unique management challenge. Ultimately, the patient underwent a re-operation after specialist multi-disciplinary discussion and is currently disease free at 18 months post-surgery. The authors wish to contribute their experiences of managing this rare extraspinal ependymoma to the few existing reports in the literature.
INTRODUCTION
Ependymomas account for 60% of spinal tumours of neuroglial cell origin [1]. They arise from the ependymal cells lining the ventricular system of the central nervous system, which are responsible for regulating the composition of cerebrospinal fluid [2]. Ependymomas can therefore be found anywhere along the neuraxis and appear to have a bimodal age presentation, most commonly occurring in the first and fourth decades [3].
Although uncommon in adults (accounting for only ~2% of all adult primary central nervous system tumours), ependymomas in this population arise most commonly in the spine. However in the paediatric population 90% of ependymomas are seen intracranially, especially in the posterior fossa [3]. Rarely, ependymomas have been reported to arise extraspinally, most commonly as sacrococcygeal tumours, and the pathogenesis of how the tumour arises outside of the neuraxis, in the apparent absence of ependymal cells, is poorly understood [1].
Just 0.5% of ependymomas are classified as myxopapillary, a rare histopathological subtype sometimes associated with a more favourable prognosis due to its usually benign nature [4]. Since they were first described in 1902, there have been only isolated reports of extraspinal sacrococcygeal myxopapillary ependymoma in the literature. Therapeutic approaches can therefore vary, particularly given the potential for local recurrence and even metastasis. We wish to report on our experience of managing an extraspinal sacrococcygeal myxopapillary ependymoma in a paediatric patient.
CASE REPORT
A 9-year-old Caucasian male presented with a 3-year history of a persistent swelling in the natal cleft. Diagnosed as a pilonidal abscess in the community, the patient had been prescribed multiple courses of antibiotics prior to surgical referral. In clinic, a soft tissue swelling measuring 50 × 30 mm was found in the natal cleft, prompting a clinical diagnosis of pilonidal cyst. There were no sinuses or midline pits, and the overlying skin appeared uninvolved. A cream-coloured solid mass measuring 50 × 35 × 30 mm was subsequently excised en bloc down to the periosteum, with no capsular breach.
Unexpectedly, histological examination revealed a large, subcutaneous, lobulated tumour with pleomorphic tumour cells (Fig. 1). The tumour cells appeared diffusely and strongly positive for S100, CD56, vimentin, glial fibrillary acidic protein (GFAP) and TFE3. Other epithelial and neuroendocrine immunostains were negative. On sections reviewed, the tumour was found to possibly extend to one of the lateral circumferential resection margins, suggesting incomplete excision. The histology was reviewed by specialist paediatric neuropathologists, who confirmed a diagnosis of subcutaneous ependymoma of myxopapillary subtype (World Health Organisation (WHO) grade 1). The patient subsequently underwent magnetic resonance imaging (MRI) of the neuraxis, which confirmed that the tumour was extraspinal in origin.
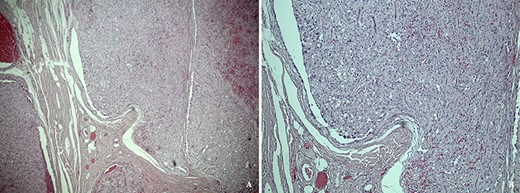
(a) Low power hematoxylin and eosin stain showing a subcutaneous tumour composed of lobules of pleomorphic epithelioid cells. (b) High power photomicrograph showing a multi-nodular nested architecture separated by fibrovascular septa, and a perivascular arrangement of cells reminscient of pseudorosettes.
Following a paediatric neuro-oncology multi-disciplinary team discussion in collaboration with the patient’s parents, a wide-local excision of the previous surgical site was undertaken. No coccygectomy or stripping of the sacral periosteum was undertaken during the second procedure as the deep circumferential resection margin from the first excisional surgery was felt to be adequate. The patient recovered well post-operatively without neurological deficit, and further histological examination of the wide-local excision specimen did not reveal any tumour cells.
Given the unusual anatomical site and morphology of the ependymoma, the option of adjuvant radiotherapy was also discussed with the patient and family. However, due to the patient’s good recovery, lack of any neurological deficit or residual microscopic disease, and the potential long-term adverse side effects of radiotherapy in a child, this was declined.
DISCUSSION
The WHO classifies ependymomas into several subtypes, and grade 1 comprises of subependymoma (generally found in adults) and myxopapillary ependymoma as found in our case. Classic and anaplastic ependymomas form WHO grade 2 and 3, respectively [5], and the majority of paediatric intracranial ependymomas are divided amongst these latter grades [6]. Immunohistochemistry demonstrates that myxopapillary ependymomas are usually strongly positive for S100, CD56, and GFAP [7], as seen in this case. Unusually for ependymoma, our case was also positive for TFE3 and molecular analysis was therefore undertaken for TFE3 gene arrangement, but this was negative for the ASPSCR1/TFE3 fusion transcript. Histopathological examination also revealed generally low mitotic activity (up to 2/10 mitotic figures per high powered field) and a low to moderate Ki-67 proliferation index of around 5%. Myxopapillary ependymomas appear to show a slight male predominance and when these tumours do rarely occur extraspinally, they are found either presacrally or subcutaneously in the sacrococcygeal region.
As pilonidal disease is typically a condition of young people, it is unsurprising that this is a misdiagnosis. In our case, differentiating from pilonidal disease was the lack of a documented inflammatory episode or additional features such as sinus formation, chronic discharge and localised sepsis. Other common differential diagnoses include lipoma, chordoma, neurofibroma and teratoma [8]. This broad differential highlights the importance of considering diagnoses alternative to pilonidal disease in persistent and refractory swelling or pain in the natal cleft [9].
There is a lack of consensus on the optimal management of paediatric extraspinal ependymomas but wide excision, often including coccygectomy, has been described with maximal safe surgical resection being crucial in successful outcome [10]. Adjuvant radiotherapy is usually incorporated with possible further resection of surrounding structures to minimise the risk of recurrence [1, 7]. In our case, after wide-local excisional surgery, adjuvant radiotherapy was offered but this was declined for reasons previously explained. As ependymomas have the potential to metastasise or recur locally, prolonged follow-up post-surgery is recommended. Our patient remains disease-free 18 months following his first resection and is currently under regular follow-up with quarterly brain and spine MRI scans.
We therefore report a paediatric case of extraspinal myxopapillary ependymoma diagnosed unexpectedly after treatment for suspected pilonidal disease, and wish to highlight the importance of considering alternative diagnoses in those with persistent pain or swelling in the natal cleft. Our patient was managed effectively with surgery alone without the need for adjuvant radiotherapy as described elsewhere in the literature, with good results at 18 months post-surgery. This makes a case for surgical management alone in specific paediatric cases where there is no neurological deficit or residual microscopic disease after wide-local resectional surgery.
ACKNOWLEDGEMENTS
None.
SOURCES OF NON-FINANCIAL SUPPORT
None.
FUNDING
None.
CONFLICT OF INTEREST STATEMENT
None declared.

{kind=link}