





Abstract
Von Recklinghausen disease (neurofibromatosis type 1—NFT1) is a genetic disorder with autosomal dominant inheritance pattern, caused by mutation of a tumour suppressor gene. Its main features include multiple cutaneous café-au-lait spots and neurofibromas. It is associated with an increased risk of developing neuroendocrine tumours, for instance, in the duodenum. The authors present a case of a 23-year-old male patient admitted to the emergency department due to persistent vomiting. Imaging and biopsy studies revealed an obstructive and large duodenal neuroendocrine tumour; hence the patient underwent a pancreaticoduodenectomy.
INTRODUCTION
Von Recklinghausen disease (neurofibromatosis type 1—NFT1) is a genetic disorder with autosomal dominant inheritance pattern, caused by mutation of the NF1 gene, which encodes the neurofibromin protein that functions as a tumour suppressor. The estimated incidence of this disease is 1 in 3000 [1]. Its main features include multiple cutaneous café-au-lait spots, neurofibromas, arterial hypertension, bone abnormalities, optic nerve tumours, Lisch nodules, larger than average head size and short stature [2]. It is associated with an increased risk of developing neuroendocrine tumours (NETs) [3].
Approximately 60% of NETs are located in the gastrointestinal tract, predominantly in the small bowel (30%) [4]. The majority of them cause no symptoms, although they may present with abdominal pain or bowel obstruction. In the duodenum, surgical resection is recommended for symptomatic NET, larger than 2 cm or associated with regional lymph node metastases.
CASE REPORT
A 23-year-old male patient with a history of asthma, NF1 and major depression presented to the emergency department (ED) due to persistent vomiting, epigastric pain, dizziness and fever in the last 2 days. He also noted some weight loss and anorexia.
He was hemodynamically normal and stable and eupnoeic. His skin and mucosae were normally coloured, but slightly dehydrated. His abdomen was flat, depressible, with tenderness on the epigastrium, without guarding. Bowel sounds were present and normal. In the right lower quadrant, there was a linear scar due to appendectomy performed 11 years before. He received intravenous fluids and a nasogastric tube for decompression.
Blood tests showed no anaemia nor leukocytosis but transaminases and C-reactive protein were slightly elevated. He had an abdominal and thoracic computerized tomography scan (CT) done in the ED that revealed a large duodenal tumour, approximately 6 cm diameter, with an associated intramural haematoma and one enlarged lymph node in the liver pedicle, 19 × 13 mm (Figs 1 and 2). There was no distant metastasis. Additionally, he underwent an upper digestive endoscopy in the ED, confirming the presence of an extensive, nodular, polypoid and obstructive duodenal tumour in its first and second portions, including the major papilla. However, biopsies turned out to be inconclusive. Later, he also had a magnetic resonance done that ruled out invasion of adjacent structures, except for the pancreatic head. Finally, he underwent an endoscopic ultrasound (EUS) with biopsies showing a duodenal NET, with diffuse expression of CK7, chromogranin and synaptophysin, and Ki-67 expressed in less than 2% of the cells.
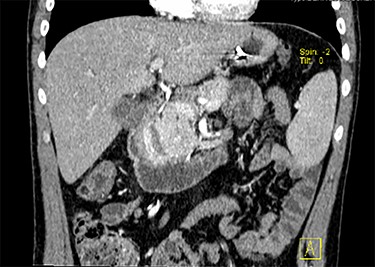
CT scan with contrast, coronal view of the large duodenal mass. Note the polypoid solid mass, in the medial border of the second portion of the duodenum, of approximately 6 cm, enhanced by contrast, slightly heterogeneous and invading the pancreatic head parenchyma. More proximally, in the right lateral border of the first portion of the duodenum, there is an extramural haematoma, well-defined, of approximately 4 × 2 cm, non-enhanced by contrast. Note also a slight dilation of the intrahepatic biliary ducts, and the main bile duct, with a progressive distal narrowing, resulting from compression of the duodenal mass.
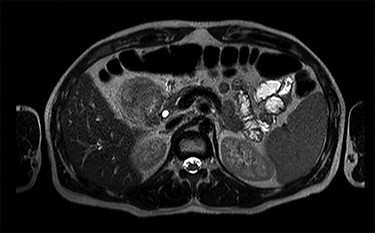
MRI scan—axial view of the large duodenal mass. Note the solid mass, enhanced by paramagnetic contrast, in the medial wall of the duodenum, invading the pancreatic head parenchyma. More proximally, in the first portion of the duodenum, there is a heterogeneous mass, non-enhanced by contrast, suggestive of an intraparietal haematoma.
This case was presented in the Multidisciplinary Oncologic Board meeting, and surgical resection was proposed. After nutritional optimization, the patient underwent a pylorus-preserving pancreaticoduodenectomy that ran uneventfully. Figure 3 shows the pancreaticoduodenectomy specimen (gallbladder not included in this image).
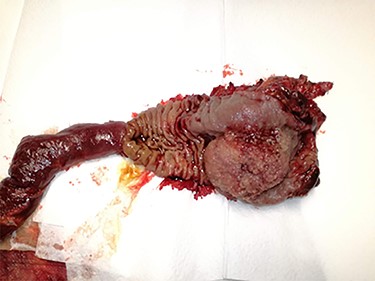
Specimen of the pancreaticoduodenectomy (gallbladder not included in this image). Note the polypoid mass of approximately 6 × 4 cm, with a pink surface, villous area, partially sectioned, invading the head of the pancreas and obstructing de major papilla.
He had an uneventful recovery and was discharged home on the ninth post-operative day.
The pathology report revealed a duodenal NET—pTNM T3N1 G2 (moderately differentiated, 5 mitoses per 10 high power fields and Ki-67 of 4.7%) that invaded the pancreatic head, with 10 metastatic nodes from the 14 analysed and multiple lymphovascular and perineural invasion. Resection margins were tumour-free. Tissue cells were positive for CAM 5.2, synaptophysin and chromogranin A.
He had a DOTANOC-PET done 2 months post-operatively that did not find residual nor metastatic disease. He remains asymptomatic (Fig. 4).
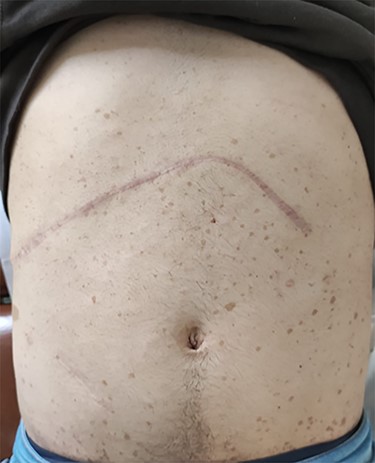
Photograph of the abdomen of this patient, showing the multiple ‘café-au-lait’ spots, on the 8th post-operative month.
DISCUSSION
This case report demonstrates that when a patient with NFT1 presents with persistent vomiting, a digestive tumour is a possibility, regardless of the patient age, especially if associated with anorexia, weight loss, low fever and asthenia.
The first approach comprises intravenous fluid and electrolyte resuscitation, and gastric decompression, followed by imaging and endoscopic studies with biopsies, and nutritional status assessment.
The NFT1 is multisystem and associated with multiple complications, such as neurofibromas, arterial hypertension, bone abnormalities, scoliosis, pathologic fractures, optic nerve tumours (gliomas), Lisch nodules, brain tumours and leukaemia [5].
The most concerning of these complications is the increased risk of developing malignant neoplasms, which share the same cellular origin—the neural crest. Besides NET, there is also an increased risk of developing GIST and pheochromocytomas [6]. Consequently, these patients should always be followed at least annually, in a specialized outpatient clinic, in order to identify early any sign or symptom suggestive of an underlying neoplasm [7].
The best studies for diagnosis and staging of NET of the gastrointestinal tract are CT and EUS, and for ruling out residual or metastatic disease, a DOTANOC-PET. This patient had an abdominal and thoracic CT scan that revealed a large duodenal tumour, without distant metastasis. Additionally, he underwent an upper digestive endoscopy that confirmed the presence of an extensive and obstructive duodenal tumour, and an EUS where the biopsies showed a duodenal NET. Finally, he had a DOTANOC-PET done that did not find residual nor metastatic disease.
Resection is the primary treatment for duodenal NET: endoscopic for lesions less than 1 cm, and surgical for those greater than 2 cm or when there is nodal metastasis [8, 9]. A tumour located in the first or second portions of the duodenum generally implies a pancreaticoduodenectomy due to the broader pattern of lymphatic drainage. Meanwhile, a tumour located in the third or fourth portions may be treated with a segmental resection.
In conclusion, NFT1 is generally recognizable for the multiple cutaneous café-au-lait spots. If a patient with NFT1 presents with persistent vomiting, we should suspect of a digestive neoplasm. NFT1 is linked to a higher likelihood of developing NET in diverse locations, including the duodenum. We show a case of a young adult patient with a history of persistent vomiting, whose imaging and biopsy studies revealed a large duodenal NET. Since resection is the primary treatment for duodenal NET, he underwent a pancreaticoduodenectomy and remains asymptomatic.
Conflict of interest statement
None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}