





Abstract
Merkel-cell carcinoma is a rare form of aggressive cutaneous cancer that is associated with a poor prognosis. Despite significant advances, its pathogenesis is still poorly understood, and treatment remains controversial. Timely diagnosis and early management is essential in improving survival rate. We present a case of a 63-year-old patient with a rapidly growing upper limb Merkel-cell carcinoma. It was treated with wide-local excision and adjuvant radiotherapy.
INTRODUCTION
Merkel-cell carcinoma (MCC) is a rare and aggressive form of cutaneous cancer that carries a poor prognosis and is associated with a considerable risk of local recurrence, as well as nodal and distal metastases.
Due to their clinical features, MCCs are frequently mistaken for other forms of cutaneous malignancy and may pose a significant diagnostic and therapeutic challenge as optimal treatment still remains a matter of controversy.
We present a case of MCC of the left upper extremity in a 63-year-old woman, treated by wide-local excision and radiotherapy.
CASE REPORT
A 63-year-old woman presented to vascular clinic with a large, painless, exophytic erythematous and centrally indurated lesion of the left arm (Fig. 1). Having grown rapidly over the preceding few months and due to recurrent episodes of bleeding, it was investigated in the community by ultrasound. Due to sonographic features of hypervascularity it was presumed to be a haemangioma and referred onto our service (Fig. 2). Her past medical history was significant for hypertension, being an ex-smoker, chronic obstructive pulmonary disease (COPD) and cancer of the left breast that had been treated with wide-local excision, axillary lymph node clearance and chemotherapy 6 years prior to this presentation.
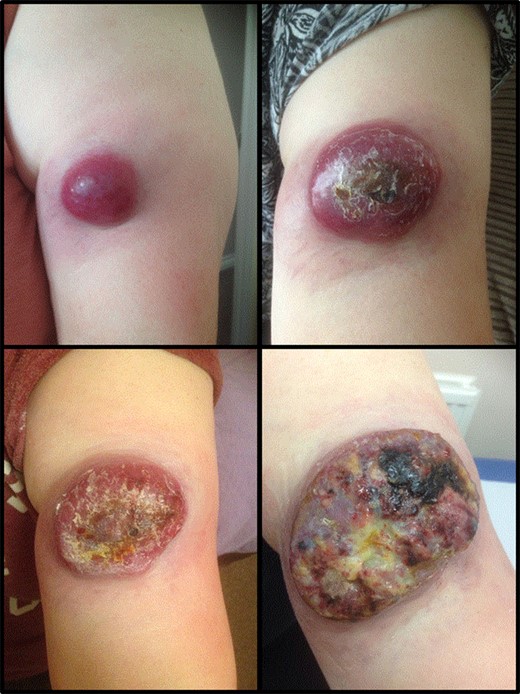
Represents the progression of the size of the cutaneous lesion over a few months.
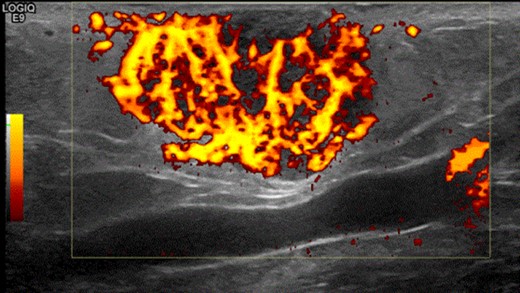
Shows the highly vascularized sonographic features of the mass on ultrasound scan.
Physical examination revealed a 12 cm × 10 cm fungating soft tissue mass with no evidence of axillary or cervical lymphadenopathy. Following urgent magnetic resonance imaging of the lesion (Fig. 3), biopsies were obtained under local anaesthesia. Histology result identified an ulcerated and invasive tumour involving the dermis (Fig. 4). It was composed of sheets of monotonous atypical round cells with hyperchromatic nuclei. The tumour cells were positive for chromogranin, CD56 and CK20 and negative for S-100, TTF-1, LCA and Melan-A. These confirmed the diagnosis of MCC and the patient was thus treated with wide-local excision and radiotherapy. Further review at 6 months confirms the patient has had a good postoperative course and is free of local recurrence and distal metastases.
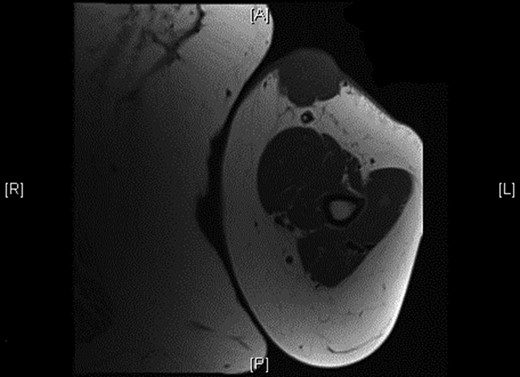
Shows the mass being superficial and subcutaneous without any underlying muscle involvement on magnetic resonance imaging.
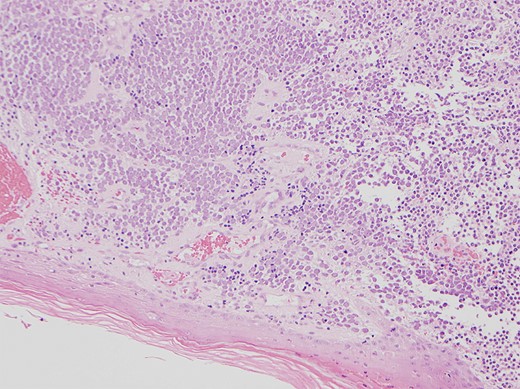
Shows the histology image of the biopsy sample with monotonous atypical round cells and hyperchromatic nuclei.
DISCUSSION
MCC, first described in 1972 by Toker et al. is a rare and aggressive form of skin cancer, believed to originate from the Merkel cells encountered at the basal layer of the epidermis [1]. It commonly presents as a painless, firm, dome-shaped, red and ulcerated lesion in sun-exposed parts of the skin. At presentation the majority are local lesions (70–80%), whilst nodal involvement (9–26%) and distal metastases (1–4%) are encountered less frequently [2].
Traditionally exposure to ultraviolet light, a history of previous skin malignancy, immunosuppression, treatment with methoxsalen (a treatment for psoriasis) and arsenic exposure have been considered risk factors for developing MCC [3]. However, a recent article highlighted the significance of the Merkel-cell polyomavirus (MCPV) in the pathogenesis and classification of MCCs [4]. Though the effect of MCPV positivity on prognosis is still not elucidated, poor prognostic factors include old age (>75 years), male gender, central distribution of the tumour (head and neck), rapid tumour growth and extension beyond the dermis [5, 6].
The optimal treatment for MCCs remains controversial, a problem that is further compounded by the rarity of this condition that precludes randomized trials, as well as its varying anatomical distribution making standardization of excision margins difficult. There is however good consensus that local excision with margins of 1–2 cm, i.e. the removal of minimal tissue, should be the mainstay of treatment [3].
An increasing body of evidence has been amassed to suggest that nodal recurrence may in fact be a delayed presentation of occult micrometastases, and thus sentinel node biopsy is advocated [7]. In our case, the patient had previously undergone axillary lymph node clearance as part of her treatment for her breast cancer, and thus sentinel node excision was not necessary at the time of her local excision.
Equally as controversial is the role of adjuvant radiotherapy. Recent reports have shown that it may improve outcomes in terms of minimizing the risk of local and regional recurrence, however, there seems to be no data to suggest that overall survival is improved [8]. Having discussed the risks and benefits of radiotherapy in the setting of a paucity of evidence in the literature, the patient opted to proceed with radiotherapy and completed it successfully.
CONCLUSION
MCC is a rare clinical entity that requires a high index of clinical suspicion and poses a diagnostic and therapeutic dilemma. Due to its rarity, there is a paucity of evidence in the literature regarding the optimal management of patients, and despite evidence to support surgery and adjuvant radiotherapy, case by case consideration is advised.
CONFLICT OF INTEREST STATEMENT
None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}