





Abstract
Phosphaturic mesenchymal tumor (PMT) is a rare mesenchymal neoplasm associated with tumor-induced osteomalacia involving bone and soft tissue that produces paraneoplastic hypophosphatemic osteomalacia. The common physiologic defect in this conditions involves an impairment in renal tubular phosphate reabsorption with a downregulation of renal 1α-hydroxylase activity, while calcium metabolism remains essentially unaffected. Microscopic features consist of spindle cells, multinucleated giant cells and calcifications embedded in a chondromyxoid matrix with variable cellularity and prominent vascularity. Approximately 95% of PMTs involve the extremities and appendicular skeleton, with only 5% occurring in the head and neck region. Localization in the head and neck is pretty uncommon, nose and paranasal sinuses are preferentially affected. Due to its rarity, the purpose of the study was to report a new case of PMT whose locations in temporomandibular joint was never reported in literature.
Phosphaturic mesenchymal tumor (PMT) is a rare mesenchymal neoplasm associated with tumor-induced osteomalacia (TIO), a paraneoplastic syndrome that manifests as renal phosphate wasting caused by elevated serum FGF23. Besides osteomalacia, the clinical presentation includes bone pain and multiple bone fractures. The tumor cells produce a peptide hormone-like substance known as fibroblast growth factor 23 (FGF23), a physiologic regulator of phosphate levels. FGF23 decreases proximal tubule reabsorption of phosphates and inhibits 1-α-hydroxylase, which reduces levels of 1-α, 25-dihydroxyvitamin D3. Overexpression of FGF23 by the tumor cells causes an increased excretion of phosphate in the urine, mobilization of calcium and phosphate from bones, and the reduction of osteoblastic activity that can lastly produce a bony tissues loss. Patients typically present with gradual muscular weakness and diffuse bone pain from pathologic fractures. The diagnosis is often delayed due to the nonspecific nature of the symptoms and lack of clinical suspicion.
The diagnosis is established by the finding of acquired chronic hypophosphatemia due to isolated renal phosphate wasting with concomitant elevated or inappropriately normal blood levels of FGF23 and decreased or inappropriately normal 1,25-OH2-vitamin D (1,25(OH)2D).
Locating the tumor is critical, as complete removal is curative. For this purpose, a step-wise approach is recommended, starting with a thorough medical history and physical examination, followed by functional imaging
Microscopic features consist of spindle cells, multinucleated giant cells (MNGCs) and calcifications embedded in a chondromyxoid matrix with variable cellularity and prominent vascularity. It contains ‘grungy’ calcification and MNGCs. Nuclear chromatin is relatively well dispersed and cell morphology is bland with minimal mitosis. Scattered MNGCs and areas of erythrocyte extravasation are also seen.
Typically PMT behave as a benign neoplasm and malignant PMTs are uncommon, rarely metastasize and only 10% recur [1].
PMT typically occurs in adults but pediatric cases have been reported with no significant gender predilection. Frequently it is discovered in the soft tissues surrounding the extremities; ~95% of PMTs involve the appendicular skeleton, with only 5% occurring in the head and neck region [2]. Historically, it has been reported that, of the PMTs located in the craniofacial region, the majority (more than 80%) are found in the nose and paranasal sinuses [3], jaws localization is exceptionally rare.
Even though very few cases of PMT have been reported in the world literature, it is very important to consider this diagnosis in all patients with hypophosphatemic osteomalacia.
Due to its rarity, the purpose of the study was to report a new case of PMT whose locations in temporomandibular joint (TMJ) was never reported in literature.
MATERIALS AND METHODS
Patient presentation
In june 2017 a 62-year-old female referred to our department complaining for a painful slowly growing swelling at her left TMJ which onset was reported 1 year before.
She had a history of long-standing progressive back pain extended to lower joints, resulting in difficulty moving which had started in 2012 and was treated in 2012 with NSAIDs with poor results. A height reduction from 168 to 163 cm was also reported by the patient.
She had already referred to a rheumatological consultation and she underwent different blood test:
Hu, Yo and Ri antibodies
Amphiphysin antibodies
Anti-CV2 antibodies
Antigen Ma2 autoantibodies
Anti-glutamic acid decarboxylase (GAD) autoantibody
Onconeural antibodies EB1
They results were all negative.
In 2014 she was given a diagnosis of osteoporosis and was placed on therapy with Cholecalciferol (1 cps/die), further questioning revealed a negative family history of the heritable forms of osteomalacia.
At the beginning of 2017 she referred to an Otolaryngology department complaining about onset of vertigo associated with tachycardia and dyspnoea.
Additional workups revealed high levels of serum calcium and the presence of high levels of thyroglobulin, with a thyroid increased in volume.
A CT scan showed degenerative alterations and deformation in the superior part of left condyle, close to external acoustic meatus.The main finding was a calcific oval-shaped formation of 3.5 cm diameter located in the left TMJ. Macroscopic features were peculiar for non-malignancy behavior. Its radiological appearance was thereby suggestive of a benignant neoformation of the articular joint (Fig. 1).
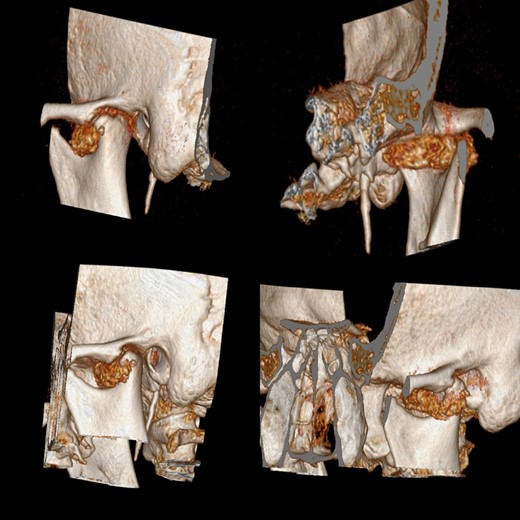
CT scan before surgery shows an oval-shaped formation of 3.5 cm diameter located in the left temporomandibular joint.
Physical examination revealed a painful swelling located right in front of tragus and difficulty in mouth movements.
Given the benignant appearance of formation we decided to perform a conservative surgical approach with resection of the tumor and sparing of the surrounding tissues.
Intraoperatively the tumor appeared encapsulated, without any erosive behavior and easily cleavable from the surrounding structures. It was located right inside the articular capsule in its superior extent and it was easily detachable from the condylar head and from the glenoid fossa (Figs 2 and 3).
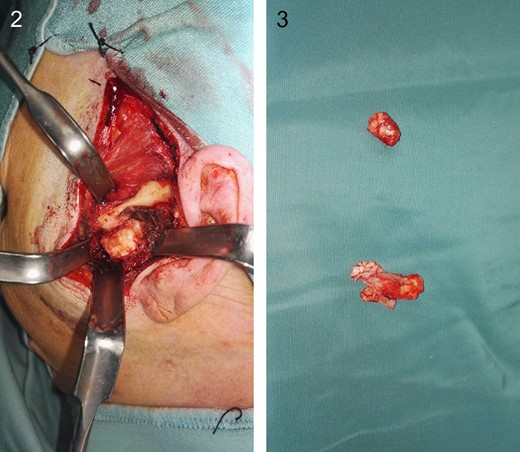
How tumor looks like intraoperatively. It was located right inside the articular capsule.
After the operation patient has recovered fast and dramatic improvement of pain and mouth movement limitations were reported.
The first histological report showed moderately atypical cartilaginous tissue, cells were resulted positive for S100 proteins and AE1/AE3 negative, the sample was then suggestive of a low grade neoplasia with atypic cells. However, no clear diagnosis was made.
For this reason our histology department asked for a second opinion from a specialized center for soft tissue cytodiagnostic and cytogenetic in Treviso, Italy. It resulted that resected tumor was characterized by spindle cells associated to calcifying particles FGF23 positive. Findings were compatible with PMT (Figs 4 and 5).
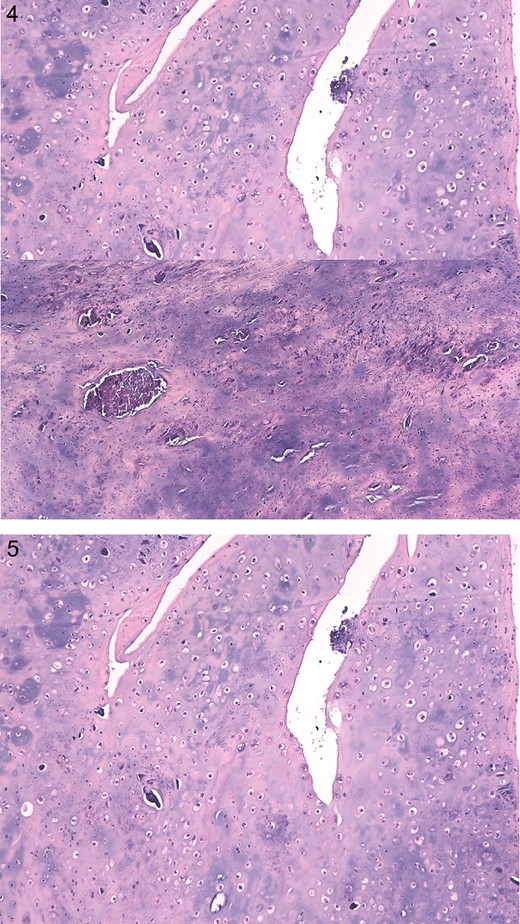
Definitive histological report showing grungy calcification and multinucleated giant cells positive for S100 proteins and AE1/AE3 negative.
After 6 months of follow-up, the patient shows no evidence of recurrence or development of TMJ pathology, vertigo, tachycardia and dyspnoea have disappeared (Fig. 6).
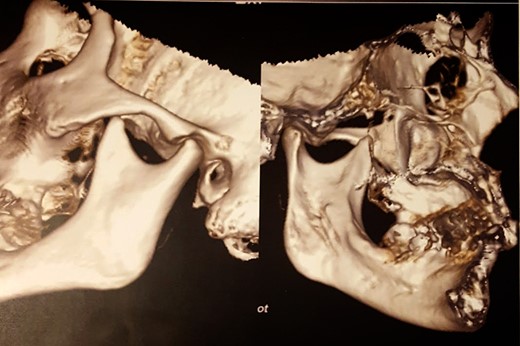
CT scan after surgery.
DISCUSSION
PMT is a rare neoplasm involving bone and soft tissue that produces paraneoplastic hypophosphatemic osteomalacia. It is most likely caused by a deficit in renal tubular phosphate resorption in which FGF23 seems to be the main responsible [4].
The biochemical derangements that characterize oncogenic osteomalacia consist of depressed levels of serum phosphate and 1,25-dihydroxyvitamin D3, together with elevated levels of urine phosphate.
The common physiologic defect in this conditions involves an impairment in renal tubular phosphate reabsorption with a downregulation of renal 1α-hydroxylase activity, while calcium metabolism remains essentially unaffected. Laboratory investigations usually show normal serum levels of vitamin D3 and calcium, distinguishing these conditions from more common forms of osteomalacia like primary vitamin D deficiency [5].
Since FGF23 secreted by the tumor causes 1,25-dihydroxyvitamin D deficiency, the tumor may indirectly causes secondary hyperparathyroidism that finally leads to osteomalacia. Progressive weakness, bone and muscle pain and pathologic fractures constitute typical symptoms.
The only possible treatment for TIO is the surgical removal of causative tumor from the body.
While these tumors are often small and not locally aggressive, they are not encapsulated and tend to be locally infiltrative.
First case of PMT was described by Weidner and Santa Cruz in 1987 [4]. Approximately 95% of PMTs involve the extremities and appendicular skeleton, with only 5% occurring in the head and neck region. Localization in the head and neck is pretty uncommon, nose and paranasal sinuses are preferentially affected [2].
In 2012 Quoting Jiang et al. reviewed 308 tumor-induced osteomalacia cases reported in literature between 1987 and 2011, among them ~46% of reported cases of TIO have occurred in females and 56% in males, with a mean age of 45.3 years. Their symptoms included muscle weakness/fatigue (100%), bone pain (100%), trouble walking (100%), reduced height (25/39, 64.1%) and pathological fractures (33/39, 84.6%), primarily in the ribs, vertebral bodies and femoral neck [6].
Within the reported cases, 56% of tumors were located in the lower extremities, 5% in the upper extremities, 3% in the hip, 31% in the head (eight in mandible and maxilla, four in nasal sinus) and 5% in the thorax region.
We found out that between 1972 and 2016 a total of 73 articles were produced regarding PMT in head and neck and 56 of them were case report. Involvement of lower jaw is extremely rare with only four cases reported in literature to date [3, 5, 7–9] and no report of cases affecting the TMJ was found in literature.
CONCLUSION
PMT is a rare mesenchymal tumor whose localisation in the TMJ was never reported [10]. This case shows that this rare neoplasm can be included in the differential diagnosis when dealing with TMJ benignant neoformations.
CONFLICT OF INTEREST STATEMENT
None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}