





Abstract
Paragangliomas (PGLs) are rare, extra-adrenal tumors, originating from neural crest cells and can occur anywhere from the skull base to the pelvic floor. Although these tumors are often benign, a fraction of malignant cases exist. Few isolated cases of malignant head and neck PGL are reported in the literature. Treatment algorithms rely heavily on retrospective case studies and institutional experience. We report an unusual case of an extensive, hereditary PGL, with invasive characteristics, that was refractory to radiation therapy. An operative approach was selected for recurrent disease in the setting of critical neurovascular structure compromise. Six months postoperatively, the patient was recovering as expected and had no evidence of recurrent disease. We propose a modified treatment algorithm based on an updated literature review that encompasses the spectrum of PGL, from benign and asymptomatic to invasive and malignant disease.
INTRODUCTION
Paragangliomas (PGLs) are rare, extra-adrenal tumors originating from neural crest cells and therefore are highly interlaced with neurovascular structures. PGL tumors migrate outside the sympathetic chain and acquire glandular characteristics [1–3]. More than 50% occur in the head and neck, especially the carotid body [4, 5]. Only 5% secrete catecholamines like their adrenal counterpart, the pheochromocytoma [1, 2, 6]. In the absence of functioning tumors, symptoms reflect compression of affected surrounding structures [3, 7]. The most common genetic associations are germline mutations of succinate dehydrogenase (SDH) via autosomal dominant transmission and carry a higher risk of malignancy [1, 3, 6]. Malignant PGL tumors are rare and defined exclusively by the presence of metastases invading nonneuroendocrine tissue [3, 5–7]. Clinical trials for PGL are scarce and often include patients with pheochromocytoma. Treatment relies heavily on retrospective case studies and institutional experience.
CASE REPORT
A 59-year-old, previously healthy woman presented to another hospital with a 2-week history of postprandial syncope and labile blood pressure. She endorsed preceding 4-month history of lightheadedness and intermittent neck pain. She recalled having a neck mass for many years and a paternal family history of benign, asymptomatic neck masses for three generations. Initially, her episodes were diagnosed as vasovagal syncope. Physical examination demonstrated bilateral palpable masses just under the angle of the mandible, originally attributed to incidental congenital anomalies and unrelated to the syncopal episodes.
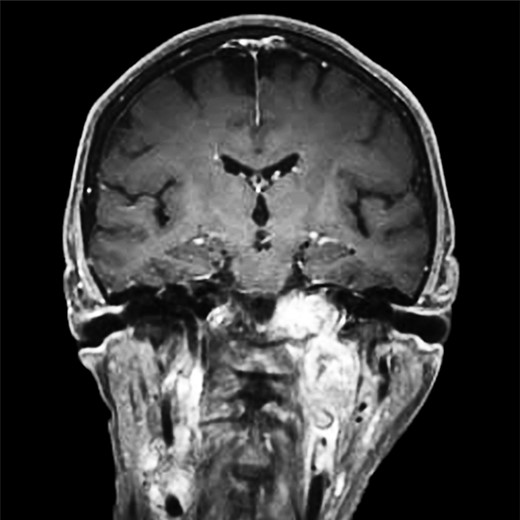
Computer tomography imaging (Fig. 1) revealed multiple bilateral PGLs extending from the carotid bifurcations to the base of the skull. The tumors were engulfing the vagus nerve, making them inoperable at the time. The PGLs were deemed nonfunctional by laboratory assessment of urine and plasma metanephrines and catecholamines. Genetic analysis revealed a SDH-D subunit mutation, an autosomal dominant mutation, resulting in markedly elevated risk of PGL tumors, especially bilaterally.
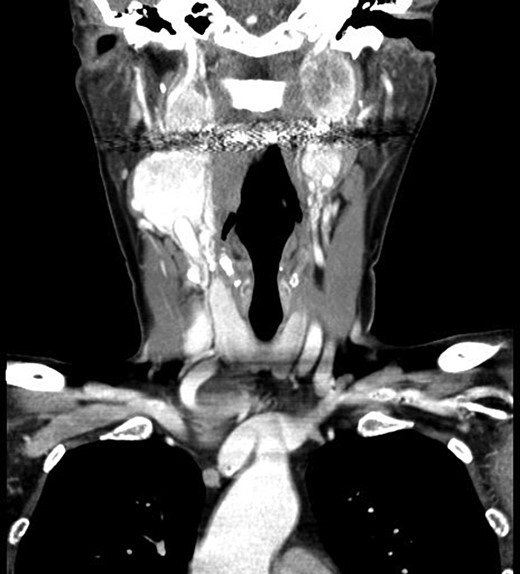
Computed tomography, coronal view, shows multiple bilateral PGLs extending from the carotid bifurcations to the base of the skull, engulfing the vagus nerve.
Radiation therapy was selected in the setting of local extension of the tumors and proximity to critical neurovascular structures. The patient underwent stereotactic radiation treatment of 30 Gy over 5 days, with a positive response on imaging (Fig. 2) and a reduced frequency of syncopal events.
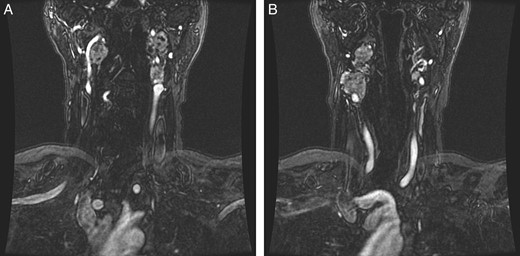
(A and B) Magnetic resonance images, coronal planes, highlight three of the four previously noted cervical PGLs decreasing in size, with the right inferior PGL remaining unchanged.
Three years after stereotactic radiation treatment, new hoarseness and dysphagia developed. Follow-up imaging showed stable disease, and thus her symptoms were attributed to post-radiation changes. Six months later, she reported new double vision, decreased hearing, worsening dysphagia and weight loss requiring placement of a percutaneous endoscopic gastrostomy tube.
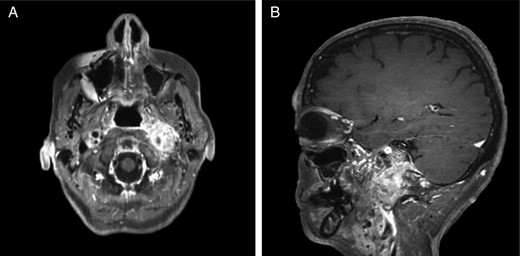
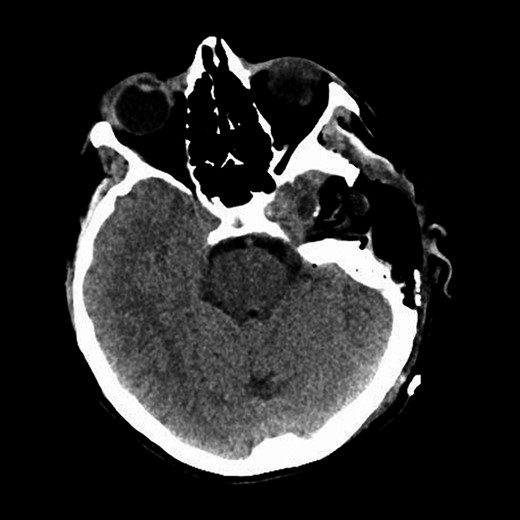
Repeat magnetic resonance imaging (Figs. 3 and 4) showed increased left-sided carotid PGL, significant extension into the jugular foramen and skull base, and associated high-grade tumor compromise of the left internal carotid artery; no radiologic evidence suggested intracranial ischemic sequelae. Computed tomographic (CT) imaging showed infiltration of the left petrous and clivus regions of the skull (Fig. 5). Findings were compatible with progression of the previously known tumor.
(A and B) Magnetic resonance images, sagittal and coronal planes, show increased left-sided carotid PGL, with significant extension into the jugular foramen and skull base.
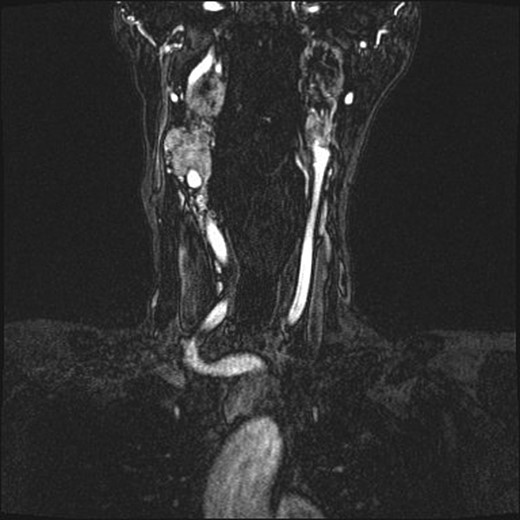
Magnetic resonance angiogram, coronal plane, shows tumor compromise of the left internal carotid artery.
CT of the head with contrast, transverse section demonstrating infiltration of left petrous and clivus regions of skull.
An operative approach was selected in the setting of progressive neurologic deficits with increasing tumor size. The patient underwent left internal carotid artery angiography (Fig. 6); vascularity of the PGL is demonstrated. The patient underwent left transjugular craniotomy with temporal bone resection, extensive resection of the left PGL, left carotid resection, cranial nerve IX, X, XI and XII resection, with anatomic preservation of the cochlea and facial nerve. In addition, intraoperative stereotaxis treatment was elected to reduce risk of disease recurrence. Postoperative CT imaging (Fig. 7) showed operative changes from the left temporal craniotomy and resection. Pathology confirmed the diagnosis of malignant PGL with positive margins.
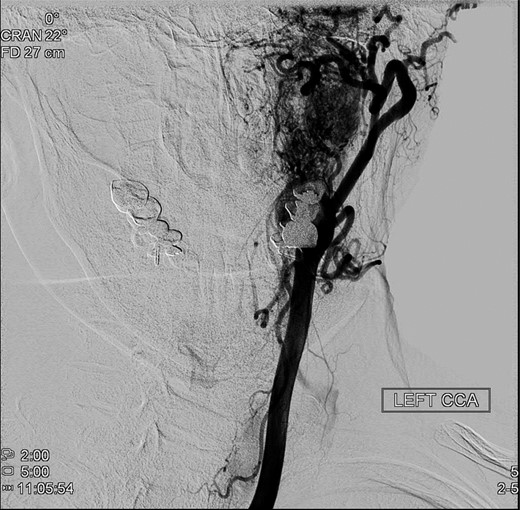
Preoperative angiography shows intricate vascularity of the PGL.
Representative CT image of the head shows postoperative changes associated with left temporal craniotomy and resection.
The patient completed all 30 planned fractions of adjuvant radiation therapy to the skull base (total dose, 6000 cGy) with curative intent. Four months postoperatively, imaging was stable without evidence of recurrence. As of manuscript preparation, the patient is undergoing cranial nerve rehabilitation and is progressing as expected, with gradual recovery of swallowing function. Interestingly, facial nerve recovery is occurring as well, despite its complete devascularization.
DISCUSSION
PGLs are rare, extra-adrenal tumors, originating from the neuroectoderm that migrate outside the sympathetic chain and acquire glandular characteristics. Overall, positive prognostic features are benign, nonfunctioning, slow-growing tumors. Anatomic location and affected surrounding structures are strongly associated with morbidity.
The most common genetic association is germline of SDH occurring via autosomal dominant transmission. The literature suggests that mutation carriers should undergo baseline magnetic resonance angiography of the head, neck, thorax, abdomen and pelvis and receive genetic counseling [4, 5].
Generally, treatment is reserved for symptomatic tumors and malignant disease. Surgical resection is well established as the only curative option. Classically, early excision has been recommended because of the unknown malignant potential of PGL and concern for progressive, local advancement over time; even in the benign setting, PGL can ultimately cause compromise of surrounding neurovascular structures [1, 6]. A recent report [6] suggested stereotactic radiation as first-line therapy, considering the high morbidity associated with open resection for head and neck PGLs.
Makeieff et al. [8] suggest that surgery should be recommended after considering the balance between the ability to achieve negative margins versus the level of morbidity that is acceptable to the patient. However, more recently, institutional practice in performing subtotal resection with observation of the residual tumor is becoming more accepted to reduce surgical morbidity. Wanna et al. [2] reported in 2014 that if >80% of the preoperative tumor volume was resected, the residual tumor was less likely to grow over the 44-month follow-up interval. Furthermore, in their population, no patient had permanent lower cranial nerve neuropathy postoperatively.
Malignant PGL disease is defined exclusively by the presence of metastases invading nonneuroendocrine tissue. Conventional, nonsurgical treatment for nonresectable, malignant PGL includes radionuclide therapy or alkylating agents. Targeted therapies have gained attention in recent years. In 2008, Joshua et al. [3] reported in Ontario, Canada, success in three cases of malignant PGL treated with sunitinib, a tyrosine kinase inhibitor. Subsequently, sunitinib has advanced to a Phase II clinical trial for patients with recurrent malignant PGL or pheochromocytoma.
We propose an updated algorithm based on review of the literature (Fig. 8) to guide individualized decision-making in the therapeutic approach for patients with PGL tumors that encompass the spectrum from benign to malignant diseases [4, 5, 7, 9].
![PGL Treatment Algorithm. Major symptomatic criteria are complete cranial nerve palsy, progressive disease with impending threat to critical neurovascular structures, intracranial extension or recurrent disease refractory to other therapies. Minor symptomatic criteria are symptomatic disease in the absence of major symptomatic criteria (e.g. syncope). CT, computed tomography; MRI, magnetic resonance imaging; PGL, paraganglioma; SDH, succinate dehydrogenase; SRS, stereotactic radiosurgery. (Data from Lieberson et al. [6], Makeieff et al. [8], Fruhmann et al. [4], Patetsios et al. [5], Rao et al. [7] and Suarez et al. [9].)](https://oupdevcdn.silverchair-staging.com/oup/backfile/Content_public/Journal/jscr/2016/2/10.1093_jscr_rjw012/2/m_rjw01208.jpeg?Expires=1788229757&Signature=coWRO027Yt4AoscVI-qxgIeNYNdNHivOvIqQfvcR-3hTltV3t6Ir00yGE5jma~v6V6gHM1ZhSTfjFtDiaHocHh15U2NP2nFz63Pxf8jwpKq8Ns~KGbtNB52m46W43P56lrH~YVbXTXomrTR8Rqp2LkuM0k5J8UAH2rkbVw8oMWJXN69CSaA9Mq6EvqJiun7Iu4bEXPySfFQItpC2GH3psLwQijCw1dSMnGrY3v19pcDuSGyy15NWaK0aeXUll4oDiyoqe37jdF16ffbC77yIyUdZdrtwsp0kizlilrjNry0-r3Y9CTliGCR2YXVQ0J~hZImghU87zReUTPw8TeqCHQ__&Key-Pair-Id=APKAIYYTVHKX7JZB5EAA)
PGL Treatment Algorithm. Major symptomatic criteria are complete cranial nerve palsy, progressive disease with impending threat to critical neurovascular structures, intracranial extension or recurrent disease refractory to other therapies. Minor symptomatic criteria are symptomatic disease in the absence of major symptomatic criteria (e.g. syncope). CT, computed tomography; MRI, magnetic resonance imaging; PGL, paraganglioma; SDH, succinate dehydrogenase; SRS, stereotactic radiosurgery. (Data from Lieberson et al. [6], Makeieff et al. [8], Fruhmann et al. [4], Patetsios et al. [5], Rao et al. [7] and Suarez et al. [9].)
CONFLICT OF INTEREST STATEMENT
None declared.
REFERENCES
Author notes
Reprints: Portions of this manuscript have been published in abstract form Society of Vascular Medicine Conference 2014: Vasc. Med. June 2014;19(3):240. Written permission has been obtained for reprint.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}