





Abstract
Paragangliomas are rare neuroendocrine neoplasms originating from the embryological neural crest. In most cases, they exhibit a benign behavior. Here we report a case of a small symptomatic para-aortic paraganglioma, which was completely removed surgically and a review of the available literature regarding the optimal follow-up of a benign paraganglioma, since no guidelines are currently available for this rare entity.
INTRODUCTION
Paragangliomas are rare neuroendocrine neoplasms arising in extra-adrenal chromaffin cells derived from the embryological neural crest. When they arise in paraganglia of the adrenal medulla, they are termed pheochromocytomas. Paragangliomas occur most commonly in the head, neck and retroperitoneum, although they may develop anywhere paraganglia are distributed [1]. They are usually diagnosed in the third to fifth decade of life and most of the tumors are sporadic [2], but may also be present as part of familial syndromes, such as von Hippel–Lindau disease, multiple endocrine neoplasia (MEN) type 2 and Carney's syndrome. Although the majority of abdominal paragangliomas are benign, 15–35% of them may exert malignant potential. In comparison with pheochromocytomas, only 1% of paragangliomas are functioning tumors producing catecholamines; thus, these tumors are rarely associated with hypertension. Clinical presentation is variable and is caused either by the mass effect, such as abdominal, flank or back pain, or by its complications, such as upper gastrointestinal hemorrhage and metastases.
In this report, apart from presenting a case of a small para-aortic paraganglioma, causing vague abdominal pain, which was completely removed by surgery, we focus on the surgeon's dilemmas regarding the optimal follow-up of the patient, since no guidelines are currently available for this rare entity.
CASE REPORT
A 50-year-old woman was referred to our Clinic from a District Hospital to receive tertiary care due to an intra-abdominal mass. The patient suffered from vague abdominal pain, which began 3 months ago. At that time she underwent abdominal ultrasound that showed the existence of gallstone disease. For this reason, laparoscopic cholecystectomy was conducted. The patient was not relieved from her symptoms, seeking, soon after the operation, again medical care. Therefore, she underwent an abdominal CT scan, which showed the existence of a small (4 cm) retroperitoneal mass, adjacent to the left edge of the aorta, at the level of the lower pole of the left kidney. Under the diagnosis of para-aortic symptomatic tumor, the patient underwent a laparotomy, resulting in the total surgical excision of a highly vascular mass, which did not invade into the surrounding tissues. There was no evidence of tumors in other organs of the peritoneal cavity. During the operation and postoperatively, blood pressure was within the normal range. The dissected mass was soft, round shaped, dark brownish, well circumscribed, 4.5 × 3.2 × 2.8 cm in size. Immunohistochemistry showed intense expression of synaptophysin and chromogranin A and the index of cell proliferation Ki67 was ∼5%. It was diagnosed as an extra-adrenal paraganglioma. Her postoperative course was uneventful and the patient was discharged 6 days after the operation. An 18-month follow-up with abdominal CT scan at 1year postoperatively and the semi-annual clinical examination showed no evidence of recurrence (Figures 1 and 2).
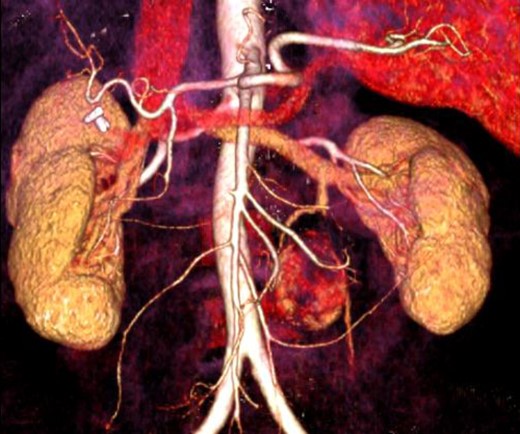
CT angiography with a 3D reconstruction.
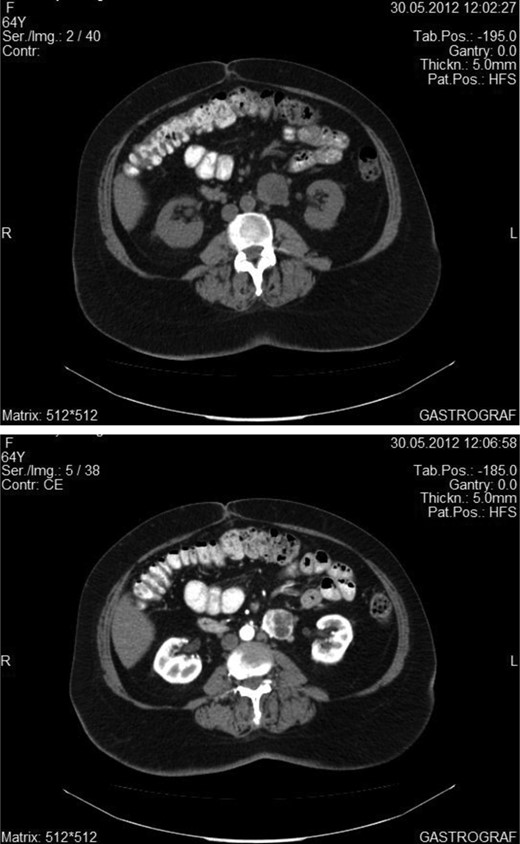
Abdominal CT showing the enhancement of the tumor after intravenous administration of an iodine material.
DISCUSSION
Paragangliomas are rare tumors representing 10–18% of all chromaffin tissue-related tumors [3]. Size is variable and >20 cm paragangliomas have been reported [4]. According to the study by Laird et al., size on its own is not correlated in a statistically significant way with malignancy in both paragangliomas and pheochromocytomas, but other factors such as invasion to adjacent organs and distant metastases are considered diagnostic for malignancy. Furthermore, genetic mutations may require more aggressive follow-up and malignant paragangliomas seem to exhibit a more aggressive behavior than malignant pheochromocytomas [5]. On the other hand, Ayala-Ramirez et al. [6] reported that not only the size of the primary tumor but also its location (especially in the mediastinum and infradiaphragmatic para-aortic area) are important determinants of both the risk for metastasis and decreased survival. Complete surgical resection is the cornerstone of treatment for paragangliomas and in the case of benign pheochromocytomas (using AFIP criteria), long-term follow-up is not necessary because the recurrence risk is very low [7]. In the case of benign paraganglioma, though, which surveillance option following surgical excision would be considered optimal?
The distinction between benign and malignant paragangliomas, as well as for pheochromocytomas, is quite difficult. Elder et al. [8] suggested the combined use of Ki-67/MIB-1 and human telomerase reverse transcriptase gene expression along with histopathology in order to identify benign abdominal paraganglioma, not at risk of recurrent or metastatic disease.
Thus, no consensus regarding optimal follow-up of benign paragangliomas exists and further data and reports relevant to the treatment of such cases are necessary for the determination of the appropriate strategy and surveillance following surgical excision of these tumors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}