





Abstract
Adrenocortical carcinoma (ACC) is a malignant tumour arising from the adrenal cortex, whereas pheochromocytoma is a tumour of the adrenal medulla with occasional presence at extra-adrenal sites. Most of the adrenocortical tumours present clinically with Cushing's syndrome and signs of virilization due to over-production of the respective hormones. It is, however, rare for an adrenocortical tumour to present clinically as a pheochromocytoma. We report the case of a 45-year-old female presenting with clinical symptoms and signs of pheochromocytoma and investigations that resulted in a diagnostic dilemma. The histopathological examination confirmed the presence of ACC after the tumour was excised. This phenomenon was due to the presence of neuroendocrine features of ACC referred to, as a pseudo-pheochromocytoma with extremely limited data in the literature.
INTRODUCTION
Adrenocortical carcinoma (ACC) is a rare malignant tumour of the adrenal cortex with an approximate incidence of 1 to 2 cases per million adults annually [1, 2]. Most of these tumours are functional, presenting clinically with Cushing's syndrome and/or signs of virilization. It is extremely rare for an adrenocortical tumour to present as an adrenal medullary tumour viz., a pheochromocytoma, thus posing a diagnostic and therapeutic challenge. Here, we report the case of an ACC mimicking a pheochromocytoma clinically, biochemically and radiologically.
CASE REPORT
A 45-year-old female presented with complaints of vague abdominal pain for 3 years. She later developed headache with episodes of palpitations, sweating and vertigo. She was diagnosed with hypertension and put on anti-hypertensive medication but failed to achieve control with regular use of the prescribed medication.
On examination, there were no signs of virilization or Cushing's syndrome. The pulse rate was 92 bpm, regular, good volume, without delay. Blood pressure (BP) was 170/100 mmHg, right arm, supine. Postural hypotension was noted with standing BP of 120/90 mmHg. Per abdomen, a ballotable mass of ∼5 × 5 cm was palpated in the right lumbar region, firm, non-tender, non-fluctuant and non-pulsatile.
Routine laboratory tests were unremarkable. Ultrasound of the abdomen showed a 5 × 5 cm solid-cystic mass overlying the right kidney. CECT abdomen confirmed the presence of a right suprarenal tumour measuring 6 × 5 × 5 cm with solid-cystic components and fluid levels suggestive of intratumoural haemorrhage. Magnetic resonance imaging (MRI) of abdomen suggested an space occupying lesion in the right adrenal gland with multiple cystic spaces of variable sizes suggestive of pheochromocytoma. Fat planes to the adjacent structures were preserved with no evidence of metastases (Figs 1–4).
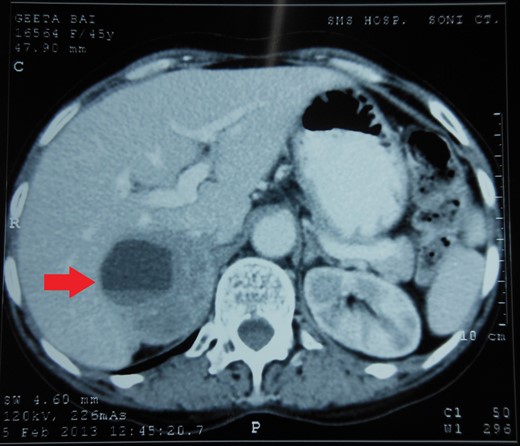
CECT abdomen showing an inhomogeneously dense right suprarenal mass (TRANSVERSE SECTION).
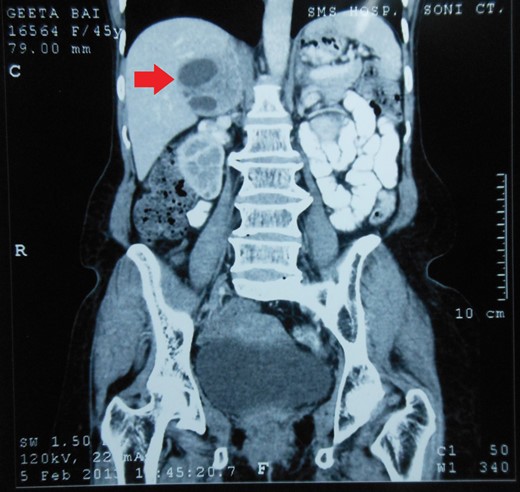
CECT abdomen showing an inhomogeneously dense right suprarenal mass (CORONAL SECTION).
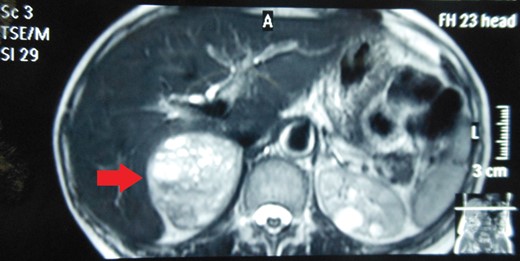
MRI abdomen showing a hyperintense right suprarenal mass (TRANSVERSE SECTION).
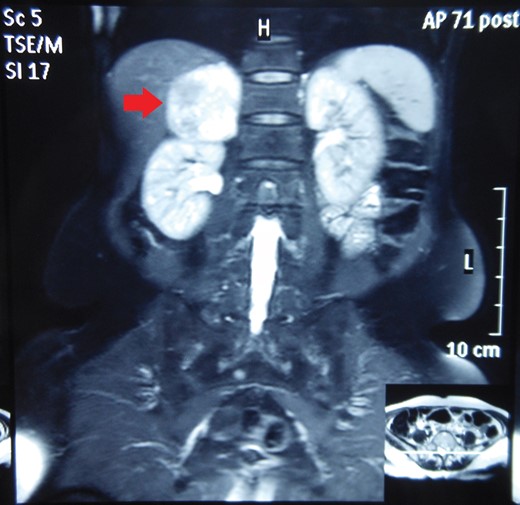
MRI abdomen showing a hyperintense right suprarenal mass (CORONAL SECTION).
Serum cortisol and 24-h urinary cortisol were normal. However, 24-h urinary vanillyl mandelic acid (VMA) was 8.8 mg (n=1.8–7 mg/24 h). Owing to limited resources and low socio-economic status of the patient, further diagnostic biochemical tests, such as fractionated plasma-free metanephrines, urinary catecholamines and metanephrines, could not be undertaken.
Pre-operative control of BP was achieved within 3 weeks with oral phenoxybenzamine 10 mg during the first week, increased to 10 mg 6 h during the second week and 10 mg 4 h during the third week. A beta-blocker was added during the second week. Volume expansion was achieved with 0.9% normal saline infusion 2 l/day during the immediate pre-operative week.
During elective right adrenalectomy, the BP shot up to 180/140 mmHg (pre-operative BP was 140/90 mmHg). Control was achieved by continuous infusion of inj. nitroglycerine and IV bolus of inj. esmolol. The tumour was found to be abutting the IVC. Post-operative recovery was uneventful. No anti-hypertensive agents were prescribed during the hospital stay or on discharge (Figs 5 and 6).
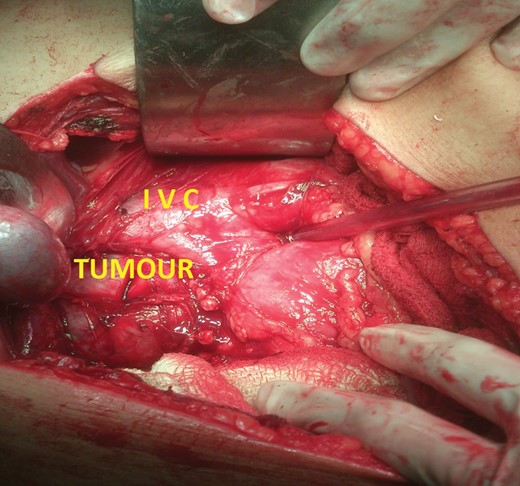
Intra-operative photograph of right adrenal tumour abutting the IVC.
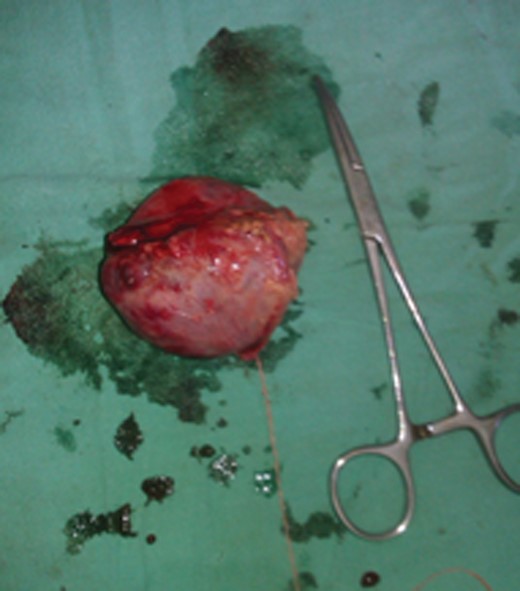
Post-operative specimen.
Histopathological analysis reported a well-encapsulated cellular lesion composed of sheets, cords and trabeculae of round to oval epithelial cells with abundant eosinophilic cytoplasm and vesicular nuclei showing Grade 1–2 pleomorphism. Foci of bizarre-looking cells with frequent mitotic figures (1–2/10 HPF) seen. Focal areas of capsular invasion were seen without vascular invasion. A histopathological study indicated ACC (Figs 7 and 8).
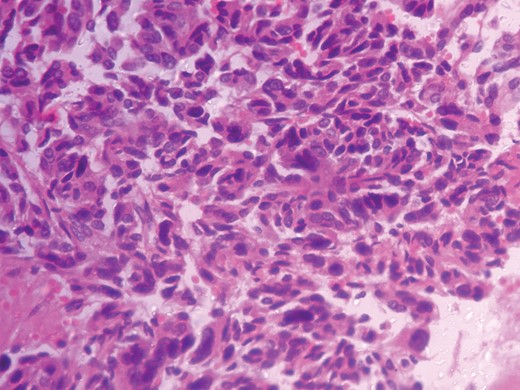
Histopathologic photograph-Low magnification.
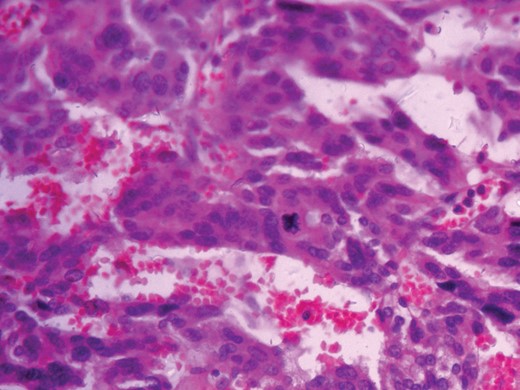
Histopathologic picture 40×.
DISCUSSION
ACC is a rare malignant tumour of the adrenal cortex with an incidence of 1 to 2 cases per million adults annually and bimodal presentation mostly during the fourth to fifth decade of life and the first decade [1, 2]. Most of these tumours are functional, and secrete glucocorticoids and androgens excessively, causing clinical Cushing's syndrome and signs of virilization, both of which can be the initial presentation [2]. They are associated with a poor prognosis [3]. It is extremely rare for ACC to present clinically as a pheochromocytoma.
Pheochromocytoma is a tumour of the adrenal medulla which secretes catecholamines. The annual incidence is ∼2–8 cases per million and presents during the third to fourth decade of life, with 10% cases occurring in children [4]. The clinical history of pheochromocytoma includes episodic headaches, palpitations and diaphoresis with severe uncontrolled hypertension. These spells are due to the excess secretion of catecholamines viz., epinephrine and norepinephrine. Depending on the catecholamine secreted, hypertension is either paroxysmal or persistent, rarely absent.
Biochemical diagnosis of ACC is done by the standard 24-h urinary cortisol excretion test and the 1 mg-dexamethasone suppression test in the presence of Cushing's syndrome and by serum adrenal androgens in the presence of virilization. Whereas, the diagnosis of pheochromocytoma can be supported by the measurement of catecholamine metabolites such as plasma metanephrines, 24-h urinary catecholamines or their metabolites such as VMA and metanephrines [5].
Radiologically, a suprarenal mass, with a non-homogenous density on CECT scans with fluid levels suggestive of intratumoral haemorrhage goes in favour of pheochromocytoma [6]. Intermediate-to-high-intensity lesions on T2 imaging of the MRI go in favour of malignant carcinomas, whereas pheochromocytomas are very high-intensity tumours owing to higher water content [7]. Another significant utility of the MRI is to evaluate the surrounding fat planes and exclude metastases. Ultrasound has less sensitivity for detecting adrenal incidentalomas and is highly user dependant.
In the present case, our patient, was in her fifth decade of life, presenting with vague symptoms such as diffuse abdominal pain with a palpable mass, episodic headache, palpitation and vertigo, with uncontrolled hypertension and postural hypotension. She had raised urinary VMA level of 8.8 mg/24 h (n = 1.8–7 mg/24 h) with normal serum and 24-h urinary cortisol levels. Though not conclusive, the raised VMA levels caused a diagnostic dilemma. Radiological investigations further supported a diagnosis of a pheochromocytoma. Pre-operative BP control had to be achieved as described. An intra-operative boom in the BP on handling the tumour further confirmed it. Histopathology gave a diagnosis of an ACC conforming to the modified Weiss criteria.
This peculiar behaviour of the adrenocortical tumours is due to the neuroendocrine differentiation, known as pseudo-pheochromocytomas [8]. Such tumours show positivity for markers such as neuron-specific enolase, and synaptophysin and negativity for chromogranin. Testing for calretinin and inhibin further increases the sensitivity and specificity in differentiating an adrenocortical neoplasm from pheochromocytoma [9]. In the literature, only 14 cases have been reported so far making these tumours a rare entity, but which can still cause a diagnostic dilemma. To the best of our knowledge, this is the only such case reported from India.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}