





Abstract
We report the case of a recurrent phosphaturic mesenchymal tumor (PHT) of the right acetabulum in a 47-year-old man with a long history of hip pain. After primary excision of the PHT, successful remission was expected due to normal phosphate levels. Over a long period, a recurrence led to destruction of the acetabulum and loosening of the back plate of the hip prosthesis. One-year follow-up after revision arthroplasty revealed normal phosphate levels, and the patient reported no complaints.
INTRODUCTION
Phosphaturic mesenchymal tumor (PHT) is a rare neoplasia associated with oncogenic osteomalacia [1], a paraneoplastic syndrome that results from the overproduction of the phosphaturic factor FGF-23. In our review of the literature, >300 cases of PHTs have been reported, but recurrence of PHT has been rarely described. We report clinical and surgical aspects of a case of PHT that recurred 8 years following resection from the right acetabulum.
CASE REPORT
In March 2003, a 47-year-old man was referred from the endocrine clinic to the outpatient center of our department with diffuse hip pain on the right side. In 1997, he had severe osteomalacia and was treated symptomatically with phosphate and calcitriol. There was no family history of any disease inducing osteomalacia, such as inborn errors of metabolism or chronic renal disease.
At the time of our investigation, laboratory tests revealed hypophosphatemia with 0.44 mmol/l of phosphate (normal range: 0.81–1.6 mmol/l), normal serum calcium (under substitution with calcitriol) with 2.6 mmol/l (normal range: 2.0–2.6 mmol/l), and low urinary inorganic phosphorus excretion with 15 mmol/24 h (normal range: 21–85 mmol/24 h).
Magnetic resonance imaging (MRI) and computed tomography (CT) scanning showed a destructive lesion, 5 cm in diameter, in the right os ilium and obturator internus muscle (Fig. 1). FGF-23 levels were significantly high and an octreotide scan was positive for radionuclide uptake in the right acetabulum. CT-observed biopsy of the right acetabulum was performed, and histological analysis revealed a benign phosphaturic tumor (PHT) of mixed connective tissue type.
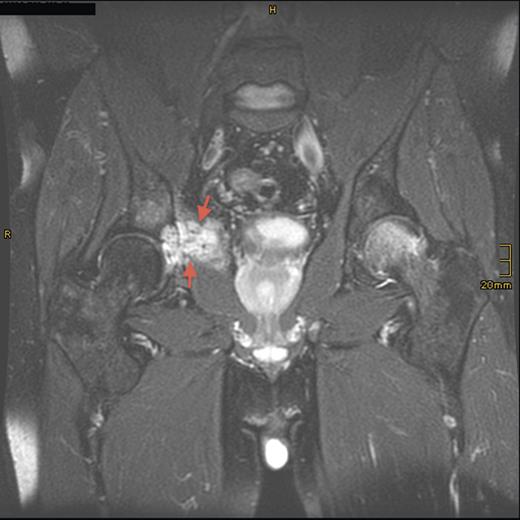
Sagittal T2-weighted, fat-suppressed contrast-enhanced MRI demonstrating the tumor lesion (5 cm in diameter) in the right acetabulum (arrows) 8 years prior to presentation.
After tumor resection through an ilioinguinal approach, serum phosphate levels normalized. To address persistent hip pain, a total hip arthroplasty was performed, and complete remission of clinical symptoms accomplished.
Eight years later, the patient presented with hip pain on the same side over a longer period. CT scan and MRI revealed tumor recurrence (Fig. 2) on the dorsomedial circumference of the acetabulum with a bone defect 2 cm in diameter. Signs of loosening of the cup of the hip arthroplasty were considered secondary. Biochemical evaluation revealed hypophosphatemia with 0.69 mmol/l of phosphate, normal serum calcium of 2.42 mmol/l and a secondary hyperparathyroidism with parathyroid hormone of 85.7 ng/l (normal range: 15–65 ng/l). An octreotide scan of the right hip showed massive enhancement of the acetabulum. Assessment of FGF-23 was inconclusive.
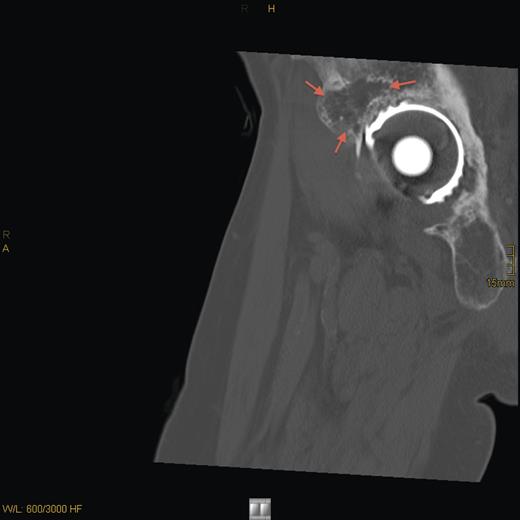
CT sagittal scan showing a suspected osteolytic lesion (arrows) in the right os ilium (8 years after primary resection).
In revision surgery, the stem was fixed but the socket was easily removed and the center of the acetabulum exhibited massive destruction. There was no clear demarcation between the tumor mass, consisting of soft tissue, and its surroundings. After wide resection of the tumor, the bone defect was filled with homologous bone and a 52-mm roof reinforcement ring (Mathys), with a cemented 50-mm low-profile cup (Mathys), was implanted (Fig. 3).
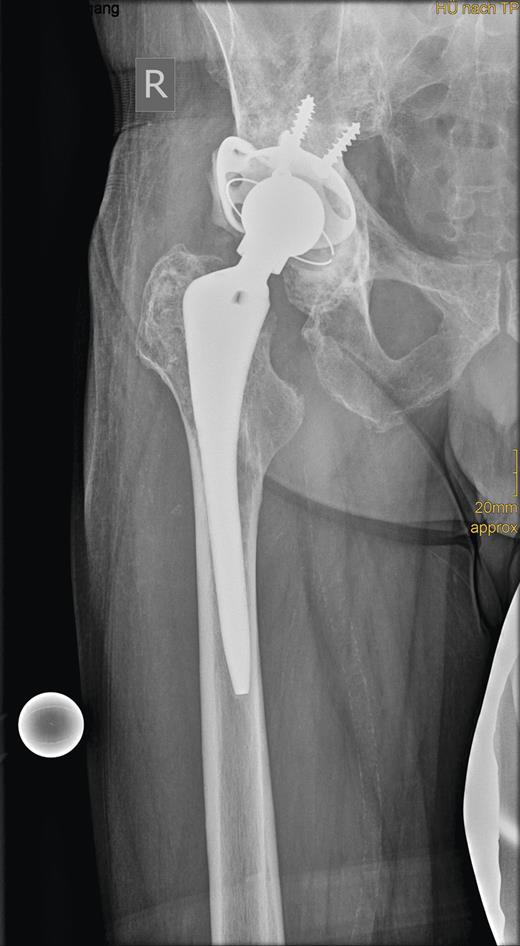
Radiograph showing the roof reinforcement ring with the cemented low profile cup after revision surgery.
Histological analysis of the removed tissue showed the same benign PHT as previously excised in 2003. Re-evaluation 1 year after revision arthroplasty revealed normal phosphate levels, and the patient reported no symptoms or complaints.
DISCUSSION
PHT is a rare lesion associated with oncogenic osteomalacia, a paraneoplastic syndrome that results from the overproduction of the phosphaturic factor FGF-23 [2–4], which inhibits renal release of vitamin D [5]. PHTs typically appear as small, benign mesenchymal tumors of various types, and are considered to consist of soft tissue or bone. In 1987 Weidner and Santa Cruz [6] published the first comprehensive study of mesenchymal tumors associated with oncogenic osteomalacia and coined the term ‘phosphaturic mesenchymal tumor, mixed connective tissue variant’. The hypothesis that these lesions comprise a single morphological entity was emphasized by Folpe et al. [7] in his consolidated analysis of 32 cases.
However, Folpe [7] and Bergwitz [8] reported the only known cases of malignant transformation of PHTs, although these tumors appeared somewhat different from the typical PHT.
PHT follows a typical course, with years of unspecific musculoskeletal pain, fatigue or muscle weakness. Reflecting the symptoms of osteomalacia, patients with PHT are commonly seen in the outpatient endocrinology and checked routinely for vitamin D deficiency or primary hyperparathyroidism. At this point, the presence of hypophosphatemia is often overlooked as previously described [8, 9]. Elevated FGF-23 levels in the blood demonstrate hypophosphatemia, although this sign is not universal, as shown by the present case. Selective venous sampling may be a useful tool; however, laboratory investigations of FGF23 should be performed in qualified laboratories using standardized protocols.
Due to phosphate wasting, patients develop secondary hyperparathyroidism leading to deregulation of calcium balance. Any other endocrine syndrome such as neurofibromatosis or inherited forms of hypophosphatemic rickets, which typically are present in childhood with a positive family history for bone and mineral disorders, have to be thoroughly excluded.
Diagnosis is complicated by difficulties in localizing the offending tumor. All previously reported cases of PHT mainly occurred in the thigh and facies crania; however, in our review of the literature these tumors have been detected almost anywhere in the body.
Suspicious areas can be identified by octreotide scanning (scintigraphy using octreotide labeled with indium-111) [9], followed by CT scanning or MRI or both as needed. Some authors even achieve exact tumor localization by co-registration of positron emission tomography and CT [10].
Symptomatic treatment includes substitution of phosphate and calcitriol; however, remission is only obtained by complete tumor resection. This aspect of treatment of PHT is complicated by the surgical problem of identifying tumor tissue that is not clearly demarcated from surrounding tissue. Primary excision of the PHT in this case led to successful remission; however, over a long period, a recurrence led to destruction of the acetabulum and loosening of the back plate of the hip prosthesis. Thus, in cases like this one, excision of the tumor mass should be wide enough to prevent recurrent disease.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}