





Abstract
Over the last 25 years, biology has entered the genomic era and is becoming a science of ‘big data’. Most interpretations of genomic analyses rely on accurate functional annotations of the proteins encoded by more than 500 000 genomes sequenced to date. By different estimates, only half the predicted sequenced proteins carry an accurate functional annotation, and this percentage varies drastically between different organismal lineages. Such a large gap in knowledge hampers all aspects of biological enterprise and, thereby, is standing in the way of genomic biology reaching its full potential. A brainstorming meeting to address this issue funded by the National Science Foundation was held during 3–4 February 2022. Bringing together data scientists, biocurators, computational biologists and experimentalists within the same venue allowed for a comprehensive assessment of the current state of functional annotations of protein families. Further, major issues that were obstructing the field were identified and discussed, which ultimately allowed for the proposal of solutions on how to move forward.
Introduction
In the early 2000s, biology entered the big data era in which all biological subdisciplines now rely heavily, both directly and indirectly, on the generation and analysis of whole-genome sequences (1), and we are still in the exponential phase of data generation (2). One of the largest benefits of the availability of sequenced genomes is the potential to elucidate the exact function of each encoded protein (3). Definitions for protein function vary and go from very broad such as the fact that a given protein is ‘an ATPase’ or ‘a transporter’, to more specific such as the protein belongs to a given protein family, as recently used in the Vanni et al. study (4), to very specific where the precise molecular function of the protein in the cell is defined. This latter definition will be the one that we are referring to in this work. The last 20 years have seen strong advances in both the generation of sequence data and the development of bioinformatic tools to predict coding sequences and regulatory elements, as well as to compare genomes and proteomes on much larger scales (5). However, the necessary functional annotations to make use of these proteomes have lost pace with other advances and have become a major bottleneck in our understanding of all forms of life (6–9). Even in the best-studied model organisms such as Escherichia coli K–12 and Saccharomyces cerevisiae, or even the minimal synthetic organism Mycoplasma JCVI-3, the latter of which contains less than 400 genes, the functions of 20–30% of their respective encoded proteins remain unknown (7, 10, 11). Although large advances have been made in the field of computational functional annotation and the manually curated subset of UniProt (Swiss-Prot) has a demonstrated error rate close to 0% for select model families, more than half of the sequenced proteome of the bacterial domain of life has no precise function (12). This is also an acute problem for Archaea (13), as well as certain eukaryotic taxa, including plants (14, 15). In general, non-model organisms remain poorly annotated with issues identified in the early days of whole-genome sequencing (16), such as limited curation resources for integrating experimental data, pollution of databases with legacy annotations, inconsistent propagation of known annotations, widely propagated errors/overprediction due to shared superfamily membership and high proportions of true unknowns remaining unresolved (9, 17, 18). Systems and synthetic biology approaches remain obstructed from reaching their full potential if the functions of biological parts continue to be left unknown or unannotated (6, 19).
Organizing a brainstorming meeting on improving protein functional annotations
To identify mechanisms to overcome this barrier, a workshop funded by National Science Foundation (NSF) (MoCeIS-DCL: Building a Network for Functional Annotation of Protein Families MCB-2129768) was held during 3–4 February 2022 at the Orlando Airport Marriott, FL, USA. The meeting was conducted in a hybrid fashion with 27 live and 32 remote participants. Six sessions, each with four short talks, were followed by breakout brainstorming groups (three live and three remote) referred to as ‘breakout sessions’ and, then, by a general debrief led by the session organizers (see Supplemental Data 1 for the full program). Two pre-meeting surveys were sent to attendees to prepare the meeting agenda (results in Supplemental Data 2 and Supplemental Data 3). Graduate student scribes participated by capturing discussions with the help of Miro boards (https://miro.com/). A Slack workspace, with a channel dedicated to each session, was used to capture interactions during the meetings and was also used as a centralized platform through which to continue post-meeting communications. All meetings, talks and remote discussions were recorded. The six sessions were articulated around two main challenges with a final goal of creating a roadmap for efficient and accurate functional annotation of the global proteome. The first challenge was how to capture, propagate and map functional information to the correct isofunctional protein subfamilies. The second challenge was determining how to change the culture among researchers, curators and developers to make the functional annotation of proteins part of the Open Science movement.
Bringing together three communities to better understand the issues at play
This meeting, one of the first that brought together three communities (biocurators, experimentalists and computational biologists) within the same venue that rarely interact in triune, showed that significant technological advances to support protein functional annotation have been made; however, these methodologies have yet to be aligned and effectively applied across databases and communities. As discussed in this meeting report, many different groups and consortia have worked to develop ontologies, create machine-readable representations of enzyme reactions and metabolic pathways, implement high-throughput (HTP) experimental methods and construct comparative genomic and modeling tools that, all together, have the potential to increase the holistic knowledge of protein function; yet many of these tools have been used exclusively for model organisms, and therein, the acquired knowledge is unable to flow between different research silos. In addition, experimentalists have not traditionally been an integral part of the biocuration cycle; because they are both providers and users of knowledge, this creates bevies of wasted time and resources. The field is at the stage where synergistic collaborative community development efforts are required to overcome the accumulated bottlenecks. The main findings stemming from the sessions’ talks and discussions are summarized below.
The capture of what is known, past and future
As a general introduction to the meeting, Valérie de Crécy-Lagard (University of Florida) summarized some of the themes emerging from the first pre-meeting survey (Supplemental Data 2). It was very clear from the survey answers that there is a major gap in functional annotation status and quality between the handful of well-curated model organisms (that have received sustained funding from NHGRI, NIH and Wellcome Trust) and the hundreds of thousands of other sequenced genomes that are dependent upon curation by specific communities or rely on automated annotation pipelines. One measure of the extent of functional annotation is the number of Gene Ontology (GO) annotations that have been curated from experimental results reported in publications. Eighty-five percent of experimental GO annotations are for genes in 10 well-studied organisms, only one of which is a prokaryote (Table 1). The second point she emphasized was the variability of functional annotations for the same protein among different databases, even for proteins that had been functionally characterized years ago (20). Experts can capture functional annotations nearly in real time in specialized databases, but this knowledge can take years to propagate across the more general resources that rely on professional curators and that are used by most biologists.
Ten most highly annotated genomes in the GO databasea
| Organism | Taxonomy | Number (%) of experimental annotations in the GO knowledgebase |
|---|---|---|
| Homo sapiens | Animals (mammals) | 145 000 (21%) |
| Mus musculus | Animals (mammals) | 123 000 (18%) |
| Arabidopsis thaliana | Plants | 70 000 (10%) |
| Rattus norvegicus | Animals (mammals) | 57 000 (8.2%) |
| Drosophila melanogaster | Animals (insects) | 53 000 (7.7%) |
| Saccharomyces cerevisiae | Fungi | 48 000 (6.9%) |
| Danio rerio | Animals (fish) | 28 000 (4.1%) |
| Caenorhabditis elegans | Animals (nematodes) | 24 000 (3.9%) |
| Schizosaccharomyces pombe | Fungi | 24 000 (3.9%) |
| Escherichia coli | Bacteria | 17 000 (2.5%) |
Only annotations with experimental evidence are reported. Numbers were obtained from the GO website, for release on 22 March 2022, excluding annotations directly to ‘protein binding’, and rounded to the nearest thousand for readability.
Iddo Friedberg (Iowa State University) then presented the notion that, due to a variety of incentives, experimentalists tend to study the same proteins again and again with little effort devoted to elucidating the functions of unannotated proteins. For example, 30 human brain proteins account for 66% of the literature. Among the incentives driving the perpetuation of the ‘ignorome’ (the set of proteins that are unannotated because they are consistently unstudied) are funding availability, technological capabilities or skills and knowledge accumulated in prominent laboratories, rather than by the biomedical or other importance of these proteins (21). Scientists tend to work on proteins that have already been characterized and that have already attracted funding (22). In summary, it appears that those proteins that generate historical interest are those that are consistently accumulating functional annotation, or, by analogy, ‘the rich get richer’ in terms of functional knowledge and the ‘poor’ are mostly ignored.
Improvement of protein databases by including chemistry
Alan Bridge (Swiss Institute of Bioinformatics) started the session dedicated to capturing functional annotation knowledge and did so by presenting on the UniProt knowledge base (UniProtKB) (23), discussing the latest advances in expert curation implemented by Swiss-Prot. UniProt is one of the main contributors to GO curation, particularly for human proteins, and his group is now using the GO Causal Activity Modeling framework, which allows GO annotations to be connected to create machine-readable models of biological pathways/networks (24). He also discussed the switch from a textual representation of enzyme and transport reactions in UniProtKB to a machine-readable format (25), which enhances the utility of UniProtKB and interoperability with other databases providing curated enzyme and transport reaction data including MetaCyc (26), KEGG (27), Reactome (28), SABIO-RK (29) and BRENDA (30). He also discussed the efforts to standardize the representation of enzyme chemistry and enzyme function in a collaboration between curators at Rhea, Reactome (31) and the GO. Finally, structure predictions from AlphaFold (32) have been integrated into UniProt for all UniProtKB/Swiss-Prot entries and those of selected reference proteomes, which will accelerate efforts to understand and predict protein functions. At the end of his talk, Alan made a case for the creation of a single Open Enzyme Reaction Database similar to the Open Reaction Database for organic chemistry (33), to which existing reaction resources such as Rhea and those cited above could contribute.
Natural language processing can improve literature capture
Zhiyong Lu (National Library of Medicine) showcased the use of natural language processing (NLP) and artificial intelligence (AI) tools to capture knowledge on protein function in PubMed under the current information overload, as two to three new papers are being deposited every second in the world’s most comprehensive biomedical literature database. LitSuggest (https://www.ncbi.nlm.nih.gov/research/litsuggest/) builds machine learning (ML) classifiers from a list of positive control papers (given as PubMed identifiers (PMIDs)) that are then iteratively calibrated, as new papers are accepted or rejected by the users (34). Another widely used tool is PubTator (https://www.ncbi.nlm.nih.gov/research/pubtator/), which performs automatic concept annotation in the biomedical literature and is particularly useful for capturing information on proteins/genes, chemical entities or diseases (35). Both PubTator and LitSuggest are already being used in production pipelines by many biocuration groups including Swiss-Prot (36), Rhea (37) and the NHGRI-EBI GWAS catalog (38). In general, AI tools required gold standard data sets and corpora to train their models. The Lu team has created a semi-automated text annotation tool, TeamTat (https://www.teamtat.org), to help create these data sets in a more automated and collaborative fashion (33).
Biocuration resources are a limiting factor
Valerie Wood (University of Cambridge), who manages the PomBase database (39), showed that mapping GO biological processes to biologically informative subsets allowed to consistently identify the percentage of proteins with unknown biological roles (process or pathway) and to distinguish these from unannotated proteins in any given genome. The number of unknown roles for the two model yeasts (fission yeast and baker’s yeast) and human has remained stable at 20% of the proteome with not much progress in the last 10 years (7). This number is similar to what is estimated for E. coli (9, 10) but changes drastically in non-model organisms as discussed by Valérie de Crécy-Lagard (University of Florida) in her introduction talk. The average in most microbes is around 50% of unknowns but may reach 70% in those less well studied (12). A recurring theme during the meeting was that biocurators are a limited resource with fewer than 100 full-time equivalents (FTE) biocurators extracting gene-specific functional information from the literature into ∼40 public databases (functional/phenotypes/interactions/pathways) and fewer than 10% of these focusing on bacteria and plants (Valerie Wood, personal communication). Community curation is often identified as one potential way to increase curation output. PomBase has a long history of soliciting author curation using incentives such as recognition in ‘research spotlights’ and promoting the use of community curation as a data dissemination activity in data management plans for funders (40). High-quality standardized curation is enabled by a user-friendly curation platform [Canto (41)], and rapid turnaround makes data visible sometimes within days of publication. Through these incentives, a quarter of the 300 000 curated ontology term assignments in PomBase are now provided by the publication authors.
Sequence embeddings can help better map proteins to families
The final talk of the session by Lucy Colwell (University of Cambridge) reported a collaboration with Alex Bateman [European Molecular Biology Laboratory, European Bioinformatics Institute (EMBL-EBI)] to show that deep learning models could be used to represent unaligned proteins as vectors that could then be used to predict the membership in a Pfam family with high accuracy (42). Interestingly, this method seems to capture different types of information than alignment-based methods, and when the two are combined, accuracy improves. This work has allowed increasing the Pfam coverage of many proteins and reveals functional similarities that cannot be detected by other methods.
Recommendations to impose good practices and standards at the publication steps and increase funding for biocuration
Two major action items emerged out of the discussions in Session 1. The first is the need to impose standards at the publication stage for protein function identification and information. Funding agencies must be encouraged to request a plan for a standardized annotation of research results—including protein functions—as part of the end of grant reporting. One could envision a knowledge management plan that could be part of the now mandatory data management plans that have to be included in proposals. The American Chemical Society (ACS) journal Biochemistry requires that authors provide UniProt identifiers for protein sequences, which facilitates the integration of literature and Findable Accessible, Interoperable and Reusable (FAIR) knowledge of protein function in UniProt and related resources (43), but they do not request functional data. A dialogue with publishers is required to develop user-friendly pipelines for authors to link literature to protein sequences and functional descriptors including ontologies such as the GO or standardized chemical structure descriptors for enzyme substrates and reactions. Schymanski and Bolton proposed a series of recommendations for the provision of FAIR chemical structure data in journals (44), which the Journal of Cheminformatics has since implemented (45). These recommendations could be easily extended to cover enzyme functions and described using universal chemical standards such as reaction SMILES, which are suitable for both human consumption and ML (46, 47).
The second is the necessity to improve functional annotation data sharing between databases, as it is particularly lacking between the generalist and more specialized databases and even between different generalist databases. Community efforts like GO have promoted the propagation of annotations, but limited funding has driven some databases (e.g. KEGG and BioCyc) to rely on subscription models that, while highly scalable, can hamper integration and reuse of their data (note that BioCyc data are made openly available after 2 years, thus enabling eventual reuse of BioCyc data). Several parallel approaches are required to solve this problem: (i) create a communication mechanism among research communities to share experiences of successful examples of functional databases (i.e. PomBase) with equivalent communities, (ii) increase biological domain-specific annotation databases for key species that can attract a specific community into a quality functional annotation effort and (iii) use the federated model to integrate databases across biological domains to facilitate communication, harmonization and interoperability. The mandates of the Global Biodata Coalition (https://globalbiodata.org/) and of Elixir (https://elixir-europe.org/) are important steps in the development of guidelines and recommendations to allow data standardization and to improve functional annotation data sharing, particularly as the framework to unify and capture information from a variety of functional databases [including but not limited to UniProtKB, GO KB, Protein Data Bank (PDB) and other more specialized resources] is largely in place. However, the number of biocurators worldwide to keep pace with information capture is inadequate by a few orders of magnitude. For example, the GO trackers have over 1300 tickets related to ontology and annotation issues but less than two FTE curators to address them, and most model organism databases have large literature curation backlogs often of up to a decade. To reach the scale needed to correctly annotate the ever-increasing global proteome, a combination of steps must occur, including an increased number of biocurators, better sharing of captured annotations between databases and increased participation of experimentalists in the curation process. Another point of discussion was the need for mechanisms for publishing negative data on protein function, which would prevent wasting resources and endlessly repeating the same functional tests. Finally, most of the proposed solutions focused on future publications, but, to capture the backlog of published functional data, several solutions were discussed. A consortium could be formed to tackle previously published literature. This could be done with different types of community annotation approaches, some relying on students such as in the Community Assessment of Community Annotation with Ontologies (CACAO) effort (48), and others relying on experts. One example of an expert-based effort is the use of annotation tools from UniProt where users can link papers to UniProt entries and provide functional information for individual entries or in batches (49). One must not underestimate the size of this task as, based on LitSuggest analyses (Alan Bridge, unpublished), tens of thousands of publications remain to be curated for enzymes in UniProt, alone. The size of this curation task is akin to what has already been captured in this database.
Identification of isofunctional families, mapping and propagating functional data to isofunctional groups
The functions of all proteins across sequenced proteomes will never be experimentally validated. The overwhelming majority of functional annotations are inferred transitively, through an operation of transferring the annotation from one protein (that has been experimentally validated) to others (that are not characterized) using some manner of determined sequence similarity. To give an order of magnitude, as of 25 March 2022, more than 64 million proteins are encoded by the ∼21 000 reference organisms in UniProt (50). Around 72 000 (or 0.1%) are directly linked to some type of experimental evidence [UniProt search terms used: ‘proteome:(reference:yes)annotation:(type:function evidence:experimental)’] (accessed × date). Hence, any information on the remaining 99.9% of proteins is inferred. Unfortunately, similarity-based methods can be error-prone by the very nature of how different functions evolve from common ancestors, and just a few mutations can change the substrate or chemistry of a given protein (51). Phylogenetic methods to transfer annotations, such as the Phylogenetic Annotation and Inference Tools (PAINT) (52) developed by the GO Consortium, have been built and are used in annotation propagation, but they are limited in the number of organisms they currently cover. Hence, a large proportion of misannotations in databases are due to incorrect identification of functionally equivalent subgroups within the same protein family (17). In the last 20 years, parallel methods have been developed to address the issue of erroneous propagation within protein superfamilies (Figure 1).
![Different existing resources to separate isofunctional families. The top three panels show different methods based on sequence similarities to try and identify subgroups. The bottom panel focuses on rule-based approaches. (A) SSN example from RadicalSAM.org with protein as nodes linked by an edge if they are similar within a certain threshold that shows the separation of the members of the Radical SAM superfamily; some subgroups cannot be separated as seen in Megaclusters 1–5; some are distinct as seen in Clusters 6–10; (B) network representation of the HIGH-signature proteins, UspA, and PP-ATPase (HUP) Superfamily (CATH 3.40.50.620) showing available functional annotations in FunFams. The colored nodes indicate FunFams annotated with different EC numbers, and the gray nodes indicate FunFams without any EC annotation, which includes nonenzymes [Figure from (127)]; (C) GO Phylogenetic Annotation: annotations of gains and losses of functions on ancestral nodes in the tree, based on experimental annotations (left) lead to different function annotations of uncharacterized proteins depending on their evolutionary history (right); (D) UniRule generation platform.](https://oupdevcdn.silverchair-staging.com/oup/backfile/Content_public/Journal/database/2022/10.1093_database_baac062/1/m_baac062f1.jpeg?Expires=1787618122&Signature=XMKKGuxbxn3LHUWY52e7HCCNlt4BFKdnSotpomb0Ub8kE1GnOEZ29eJaBT4odY6dxHu4z-88JEXMxjABJS8MGsCx2b2AGandoJYElXlgyG5f3JMwWld42C-5YLPy7ort-mK4pqCj-v-pOXo42uhwRwnLpprgL-PTEpR24VGdj3bLYJavceyU~UUys5GREqIExdbXau9bMzdOgtAA9TxL-r1kkgHhsnTR7qsQQwcKX9JXFi44NR8LoO~vt01n-0SGKHSZT-N~1W5zztck7NZosI2t2AoYQF924IFazjlrme7ZDLt3tY3MqlYIJ3CTm9a-stCZTmITOYPWJS-C2mk1Gg__&Key-Pair-Id=APKAIYYTVHKX7JZB5EAA)
Different existing resources to separate isofunctional families. The top three panels show different methods based on sequence similarities to try and identify subgroups. The bottom panel focuses on rule-based approaches. (A) SSN example from RadicalSAM.org with protein as nodes linked by an edge if they are similar within a certain threshold that shows the separation of the members of the Radical SAM superfamily; some subgroups cannot be separated as seen in Megaclusters 1–5; some are distinct as seen in Clusters 6–10; (B) network representation of the HIGH-signature proteins, UspA, and PP-ATPase (HUP) Superfamily (CATH 3.40.50.620) showing available functional annotations in FunFams. The colored nodes indicate FunFams annotated with different EC numbers, and the gray nodes indicate FunFams without any EC annotation, which includes nonenzymes [Figure from (127)]; (C) GO Phylogenetic Annotation: annotations of gains and losses of functions on ancestral nodes in the tree, based on experimental annotations (left) lead to different function annotations of uncharacterized proteins depending on their evolutionary history (right); (D) UniRule generation platform.
Challenges of separating protein families into functionally equivalent subgroups
Session 2 focused on the identification of isofunctional families. A talk from John Gerlt (University of Illinois) showed how sequence similarity networks (SSNs) can be combined with genome neighborhood diagrams to separate families into isofunctional subgroups (53) (Figure 1A). This requires cycles of increasing similarity cutoff changes and subsequent analyses by the user to choose cutoffs that are case-dependent and involve much trial and error. Recent developments of the tools show how a precomputed analysis of the Radical SAM family, one the most chemically diverse superfamilies studied to date, can greatly facilitate the correct annotation of Radical SAM subgroups with known functions as well as guide the characterization of the ones that remain to be discovered (54). Current issues of scale limit the systematic use of precomputed SSNs for all protein families, but sequence-embedding tools that do not rely on exhaustive pairwise comparisons were discussed or presented by three workshop participants: Claire McWhite (Princeton University), Christian Dallago (Technical University of Munich) and Lucie Colwell (Cambridge University). Such embeddings could solve the scalability problem, and follow-up analyses triggered by the meeting discussion are underway to explore this avenue.
Christine Orengo (University College London) presented the FunFams platform (55) and its recent improvement by integrating information on the multi-domain composition of proteins. FunFams are based on the CATH evolutionary classification that combines structure and sequence to group proteins into Mega, Super or Functional families (56) (Figure 1B). Approximately 40% of protein domains in CATH can be assigned to a FunFam having at least one experimentally characterized relative. The majority of FunFams are functionally pure, but analysis of the distribution of Enzyme Commission (EC) numbers within highly populated FunFams shows that the separation between families can still be improved and that use of sequence embeddings allows a better separation (57). Plans to integrate structures from AlphaFold 2 (58) in CATH were presented and led to an active discussion among attendees on how good structures predicted by AlphaFold could be used to dock/predict substrates with the final consensus being that it was as variable as with experimentally solved structures, mainly depending on the presence of a bound ligand in the template structure.
Paul Thomas (University of Southern California) discussed the GO Phylogenetic Annotation Project (59), which uses phylogenetic trees from the PANTHER protein family database (60) to create explicit models of protein evolution for the propagation of functional annotation of experimentally validated proteins captured by the GO Consortium (61) (Figure 1C). The strength of this system is in its use of models’ functional divergences within a protein family, identifying clades in the family with different functions, such as different substrate specificities of enzymes. Although the PANTHER trees include proteins from 142 organisms sampled across the tree of life, the focus of this effort has not necessarily been on protein families that contain members with experimentally validated GO annotations. It is therefore currently biased toward eukaryotes in general, vertebrates in particular (Table 1). This effort has been relatively successful, with 9000 families in PANTHER annotated with models of protein function evolution, covering 90% of human protein-coding genes. With the pipelines in place and many years of experience applying these tools across primarily eukaryotic species, the time seems right to expand the PAINT pipeline to include more prokaryotic species. This would require a concerted community effort to capture more experimental GO annotations for prokaryotic proteins (62) and add many more bacterial/archaeal species into PANTHER trees, as only 43 are included in the current set.
Maria Martin (EMBL-EBI) presented the UniRule system used to annotate entries in UniProt by combining InterPro signatures and taxonomy to generate annotation rules with unique identifiers (Figure 1D) (63). To date, over 8000 rules have been created and have allowed to automatically annotate half of the proteins in UniProtKB/TrEMBL. She emphasized during her talk that two points were recurrent themes throughout the whole session. First, the generation of the rules was limited by the manual capture of the experimental data to create the gold standard data set in UniProtKB/Swiss-Prot. As Valerie Wood and Peter Karp stressed in the session discussion, biocuration is massively underfunded and funding continues to decrease with only ∼3 FTE prokaryotic biocurators at UniProt, 2.5 FTE at EcoCyc, and 2.5 at BioCyc—the three main resources curating microbial function—tasked with capturing all prokaryotic functional data from the literature. Currently, the cost of curating a paper ($200–$300) is much less than the open-access publication fee for that paper (64). The second important point was that combining different methods should greatly improve the accuracy of functional annotation. She proposed to combine UniRules with PANTHER-based trees, or with FunFams, particularly in cases where the current systems fail, such as loss of function situations or species-specific moonlighting.
The major recommendations that came out of Session 2 discussions were to (i) create a publicly available set of precomputed data on protein families (like SSNs and phylogenetic trees) to save other researchers’ time and provide a shared community resource for identifying isofunctional groups; (ii) create an equivalent of a dictionary for isofunctional families to enhance findability; (iii) build online tools that can engage a large community in functional annotation tasks beyond model organisms, using similar strategies to those that have proven successful model organism databases [e.g. Pombase (34), WormBase (65) or FlyBase (66)] and (iv) greater biocuration capacity.
Propagation of functional annotations, challenges and breakthroughs
Session 3 concerned the propagation of functions between members of a protein family as well as between databases. The session began with a talk by Gaurav Moghe (Cornell University) that laid out the challenges of functional propagation from an experimentalist’s perspective using BAHD acyltransferases—a large plant enzyme family—as an example (67). He discussed how different considerations such as differences in substrate preference between duplicate genes, promiscuity/multi-functionality, context (condition/tissue) dependency of protein function, varying selection on homologs in an orthologous group and structural features like intrinsic disorder can influence the accuracy of functional prediction transfer between homologs.
Francoise Thibaut-Nissen (National Library of Medicine) then presented the National Center for Biotechnology Information (NCBI) Prokaryotic Genome Annotation Pipeline (PGAP) that is used to annotate all prokaryotic genomes in RefSeq regularly. The functional annotation is performed using a combination of domain architectures and Hidden Markov Models from PFAM, TIGRFAM or NCBIFAM, as well as BlastRules. To date, over 230 000 RefSeq genomes have been annotated using the PGAP pipeline, and product names from over 15 000 protein family models have been propagated to >80% of RefSeq proteins (68). GO terms are also being integrated into the propagated RefSeq annotations, and future models will integrate genome context.
Peter Karp (SRI International) then presented the BioCyc Web portal, comprising 19 000 Pathway/Genome Databases including 60 curated ones (69). BioCyc databases include genome, protein, reaction, pathway, metabolite and regulatory data, with the curated databases prioritizing the curation of protein and pathway data. He discussed an ‘inverse approach to functional annotation’ where, instead of predicting functions for identified genes, one first predicts functions that are likely to exist in an organism and then finds genes to associate with those functions. Four strategies for this approach, focused primarily on prokaryotic systems, were presented: using growth data under different conditions, finding transport and pathway inconsistencies, pairing orphan protein subunits with function and studying metabolic pathway ‘holes’ that can be identified and filled. He also reminded the audience that around 900 EC numbers have enzyme activities but no associated gene, a status that suggests they could be solutions for some of these ‘orphan functions’.
Finally, Christian Dallago (Technical University of Munich) discussed work from the Rost laboratory on using techniques derived from NLP modeling to represent sequences as embeddings that can be compared and are as efficient (or even more efficient than) similarity methods to transfer GO annotations (70). Two tools—the PredictProtein (https://predictprotein.org) (71) and Protein Embeddings (https://embed.predictprotein.org) servers (72)—that use the embeddings approach for predicting structural and functional properties of proteins were noted. The speed and power of this technique generated a lot of excitement and discussions throughout the meeting.
A wide range of measures are needed to improve the propagation of functional annotations
Several major points emerged from Session 3 discussions. First, it was suggested that, in addition to more biocuration and improved computational method development, the generation of more experimental data in different species or uncharacterized sub-clades of protein families would result in a lesser need for long-ranging propagations between evolutionary distant proteins and may improve prediction confidence. For example, ∼50% of the orthologous groups of BAHD acyltransferases conserved across all land plants have no characterized members (73), making their functional prediction challenging and error-prone. Generating such data, however, is a challenge, especially in multicellular eukaryotes. National labs or centers could be tasked with developing in vitro functional assays, assembling substrate libraries, prioritizing target families/subgroups of unknown function and soliciting community participation, like the Joint Genome Institute (JGI) Community Science Program.
Second, it is recognized that the many methodological advances currently happening with AI and ML are creating many new opportunities to improve functional understanding and propagation, particularly when combining ML with mechanistic modeling approaches. Alphafold is an example of this, combining classical mechanistic folding methods and techniques with deep learning to greatly improve the speed and accuracy of structure prediction. In turn, these predicted structures provide us with a new dimension of information to use when propagating functions. Similarly, the prediction of phenotypes with ML, followed by the evaluation of consistency between propagated annotations with those phenotypes through mechanistic modeling, offers another potential opportunity to enhance annotation propagation with ML and modeling. To enable classification learning, especially in multifunctional families, it was suggested that negative data from experiments should also be more systematically captured with specific sets of rules in addition to positive data, since, if an activity is lacking in a database, it is not clear whether the missing data are due to no assay having been performed or because the activity is demonstrably absent in that isofunctional group. In some cases, such negative data can be inferred automatically using existing biochemical knowledge or using taxonomic constraints (74). Further, it is vitally important that ML and modeling approaches for supporting annotation be applied almost continuously in real time, so curators and experimentalists receive rapid feedback about the impact of potential problems with their work, an effort in which KBase is actively engaged (75).
Third, we discussed whether it is feasible to produce a unified functional descriptor for a protein of unknown function by integrating available evidence, sometimes scattered across different databases. For example, is it possible to predict that ‘Protein A in a given plant species has a 90% chance of performing glycosylation of zeatin in roots under heat stress’, without actually performing the experiments in that species? Potential challenges for developing a unified model include the dependence of function on the cellular context (which can change between orthologs and paralogs), issues associated with inter-database functional transfer such as missing and unreliable information and the unresolved taxonomic scope of protein function (how far evolutionarily can we transfer the function between homologs?), inconsistent naming and specificity of function descriptions. In the latter case, without sufficient indication of their quality, it may be difficult to differentiate between accurate and imprecise/incorrect annotations, complicating the integration of different evidence into a singular functional descriptor. Integrative models like the Integrated Network and Dynamical Reasoning Assembler (INDRA) framework (see Session 4 below), which is based on graph-based analysis of structured databases and NLP, attempt to address some of these challenges (76) and produce structured descriptors of protein activity. An alternative is a ‘database of databases’ approach where users can go to a single database that displays annotations existing for a given protein in different databases of relevance, perhaps obtained using different strategies. Such an approach can reduce the need for the integration of disparate data sets into a singular descriptor and allow for varying levels of functional resolution for different protein families. Nonetheless, to enable both approaches to operate in an automated/semi-automated manner, consistent data provenance, gene IDs, data-sharing frameworks and key words are needed. The BioPAX standardized language for sharing pathway data (77) is an example in this regard.
Confidence scores should be transparent, be generated automatically with objective criteria to avoid increasing the workload for biocurators and should indicate the source, background models and extent of propagation, so that nonexperts can be more critical. Tools like ML and metabolic modeling can be applied to continuously test annotation propagations for consistency with (i) observed phenotypes (e.g. does propagating that annotation cause an organism to grow in conditions it shouldn’t), (ii) available omics data (e.g. does the propagated annotation agree with observed expression patterns or Tn-Seq insertion frequencies) and (iii) observed biological and evolutionary patterns (e.g. does the propagation place a function in a completely unrecognizable chromosomal context or a completely inconsistent taxonomic group). Even if quality control strategies are already in place for the propagation of annotations in several databases (52, 78–80), they could be further improved and more efficiently automated with the use of models. Indeed, models have an incredible capacity to rapidly and automatically integrate vast amounts of knowledge, rapidly testing (and correcting) new proposed inferences (81, 82); ML has an incredible capacity for pattern recognition, which includes recognizing inferences that fit these patterns and inferences that do not (42). Models can operate effectively in data-sparse environments (because they leverage mechanistic understanding) (83, 84); ML excels in deeply complex data-overwhelming settings (85, 86), and thus, these two technologies are deeply complementary. Applied in concert, these approaches offer a means of globally evaluating all annotations, judging for consistency system-wide. We simply need frameworks that integrate data with models in a tool that makes this kind of analysis possible. Importantly, we need to increase the number of active biocurators in the workforce to check the predictions generated by these models, particularly in the initial training stages; otherwise, errors will just propagate more quickly.
Building a new data-driven biological culture & automation of functional data generation and capture
The last 5 years have seen a global movement to adhere to good practices for scientific data management and stewardship that have been summarized as the FAIR principles (87). In the first talk of Session 4, Elisha Wood-Charlson (Lawrence Berkeley National Lab) revisited these principles with a focus on functional annotation and omics data, discussing how both incentives and mandates can be used in combination. The pressure imposed by funding agencies to follow FAIR principles is steadily increasing as data management plans are now mandated in proposals (88) and tools to help design them have been adopted by most academic institutions (https://dmptool.org/). Agencies are also providing data depositories for the research they fund (see https://science.osti.gov/Initiatives/PuRe-Data/Resources-at-a-Glance for Department Of Energy (DOE), https://www.nlm.nih.gov/NIHbmic/domain_specific_repositories.html for NIH). This is a start, but Wood-Charlson emphasized that the whole research ecosystem, from publication to promotion, has to require and reward FAIR practices and that hiring data management specialists in large teams and institutions can help ensure their implementation.
Geoffrey Hutinet (University of Florida) then discussed the challenges that database proliferation and the absence of unified protein identifiers created when teaching the use of bioinformatic tools to a variety of novice biologist users. Educators spend a lot of time drilling into students the notion that the information in databases can be obsolete or wrong and that functional data needs to be verified by cross-referencing several databases and by checking recent literature. The plethora of available databases also poses a challenge that could be eased by unique identifiers or better mapping between databases. Finally, databases need to be designed as intuitively as possible, particularly at the initial stages of interaction. Clear help or tutorials are indispensable; otherwise, users (students or professionals) will resort to other methods/tools if unable to scale the new learning curve after only a few minutes.
Stephen K. Burley (Rutgers University, Research Collaboratory for Structural Bioinformatics PDB (RCSB PDB)) gave an overview of the history of the PDB, which recently celebrated its 50th anniversary (89). Since its inception, PDB has embraced the FAIR principles emblematic of responsible data science. More recently, this commitment was officially recognized with Core Trust Seal Certification (https://www.coretrustseal.org/). PDB is a testament to how adherence to the FAIR practices can benefit both the scientific and broader communities. By defining and implementing fully machine-readable data standards, PDBx/mmCIF has been adopted by the biostructure community, making experimental 3D structure data public domain and interoperable using data exchange APIs (90, 91). Making all the information open access and available without limitations on data usage, even for commercial users, has facilitated the discovery and development of many small-molecule and biological drugs (92). Open access allowed PDB to become the reference structure database for fundamental biology, biomedicine, energy sciences and bioengineering/biotechnology (Figure 2A), literally traversing the life sciences from agriculture to zoology. PDB data are also made available for outreach and education formats by the PDB101.RCSB.org website (93). Finally, Burley emphasized that the recent turning point in computed structure modeling with the development of AlphaFold2 (58) was built on years of work by many groups that all relied upon open-access PDB data. He concluded by cautioning that users of computed structure models need to be educated as to how the reliability of these structural models can vary greatly, even between different regions of the same polypeptide chain.
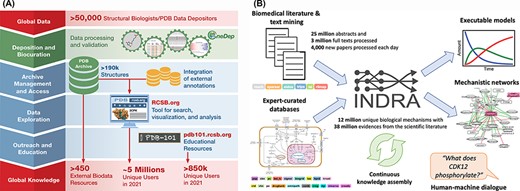
(A) RCSB PDB converts global data into global knowledge. (B) INDRA performs knowledge assembly from the biomedical literature and expert-curated databases into a knowledge base of mechanistic statements that can be converted into models and networks and queried through human–machine dialogue.
Benjamin Gyori (Harvard Medical School) discussed how automated knowledge assembly—which combines structured sources with text mined extractions from literature in a principled way—can greatly help functional annotation through the creation of structured knowledge bases that can be queried programmatically or through human–machine interfacing. Challenges in this process include the recognition of biological entities that have many different but equivalent names, as well as normalizing redundant entries that represent the same entity in different ontologies. To address these challenges, his group developed the Gilda Entity Normalization Service (http://grounding.indra.bio) (94) and Biopragmatics Stack (https://biopragmatics.github.io), a set of resources to manage bio-ontologies and their relationships (94). He then presented INDRA, a software platform that automatically assembles biochemical mechanisms extracted from the literature and pathway databases into knowledge bases and explanatory models (Figure 2B) (95). For example, using this platform on a set of protein kinases with few known substrates [often called dark kinases (96)] allowed identifying previously missed kinase targets (97) and enabled the creation of a self-updating portal for deubiquitinating enzymes (https://labsyspharm.github.io/dubportal/) (98). He also described the BioFactoid platform aimed at leveraging author input to create machine-readable pathways at the time of publication (99).
One major issue that came out of the Session 4 discussions was that large databases often already follow most FAIR principles even if they are not yet always totally open source with no systematic protocol to copy the whole database by outside users. However, the protein function information reported in papers does not. There are still papers today that publish information about proteins without providing an identifier. As pointed out in Session 1, above, clear standards imposed by publishers and, thusly, added to checklists by reviewers would contribute to solving this issue. The InChIKey representation of molecules, which generates unique alphanumeric string identifiers for chemicals (100), is an example of where FAIR standards and interoperability have already been implemented and revolutionized the ability to query databases with chemical entities once the standard was universally adopted.
Many discussions focused on the value of small expert databases that can capture very valuable curated knowledge but might not have the resources to follow FAIR practices. It was suggested that larger databases should provide guidelines and standards that the more niche databases could use, both, to allow information flow and to make possible data integration in cases where the smaller databases can no longer be maintained, as most databases have quite a short life span (101).
Finally, the constant opposition demonstrated between the machine readability and human readability for functional annotation information was discussed. Experimentalists sometimes struggle to use GO term identification for describing a function of interest as terms are not always named intuitively or have yet to be created.
Challenges in designing, conducting and capturing HTP functional screening assays to increase functional knowledge at pace with the potential of computational methods’ processing and propagating of the same knowledge
Session 5 focused more specifically on how to fill the functional knowledge gaps for the true unknowns, as opposed to fixing uncaptured or wrongly annotated problems discussed in the previous sessions. Crysten Blaby-Haas (Brookhaven National Laboratory) focuses on understanding plant protein function by combining different types of data and sequence relationships to inform function. During her talk, she emphasized that, with an analysis performed on the AmiGO2 platform (http://amigo.geneontology.org/amigo), only 0.1% of microbial proteins and 1% of eukaryotic proteins were found to be associated with at least one experimentally supported GO term, all with a clear bias toward proteins of the human-pathogen sphere. To tackle this immense gap in knowledge, Blaby-Haas advocated for the use of multiple types of comparative genomic evidence and, further, that HTP assays should be better integrated to support computationally propagated functional annotations, in addition to being used to generate actionable hypotheses for genes of unknown or uncertain function (Figure 3) (102, 103).
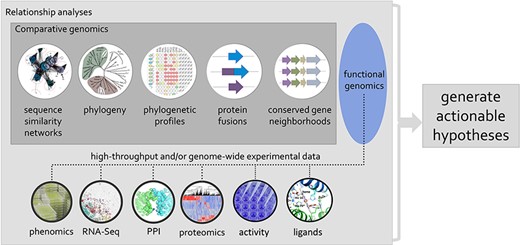
Using phylogenetic relationships to guide the integration of data associated with related proteins, mining of genomic and post-genomic data can seed defined hypotheses for the discovery of molecular and biological functions associated with genes/proteins of unknown or uncertain function.
Irina Rodionova (University of California, San Diego) presented the Palsson Laboratory Platform that dissects bacterial regulatory networks using Independent Component Analysis to identify independently modulated sets of genes called iModulons and the transcriptional regulators that control them from expression data (https://imodulondb.org/index.html) (104). This has been a powerful tool to identify which genes are regulated by regulators of unknown function or to identify the function of ‘unknowns’ under the control of known regulators. For example, the iModulon approach has allowed the prediction and subsequent validation of the unknown E. coli gene, ydhC, which was determined to encode a purine transporter (105). Future developments that will integrate iModulon with flux balance analysis models are expected to make the platform even more powerful.
Gloria Sheynkman (University of Virginia) discussed the complexity of the annotation of protein isoforms in eukaryotes that result from alternative splicing. Indeed, it is well established that different isoforms can have different functions, but, to date, less than 1% of human isoforms are annotated (106, 107). Sheynkman reported an HTP study of 366 different isoforms from 161 genes that showed that these isoforms can have wide-ranging differences in protein interaction profiles (108). The arrival of long-read RNA-seq has revolutionized the identification of the isoform field (109), and a community has been created to evaluate tools and establish standards, such as those seen with the recent Long-Read RNA-seq Genome Annotation Assessment Project (110, 111). These long reads also allow for the delineation of transcript isoforms, and, thus, allow for the prediction of full-length proteins, which enables MS-search-based detection and experimental validation of the isoform at the protein level (110, 112). Sheynkman finished by discussing the challenges of annotation for not only isoforms but for all proteoforms, including post-translational modifications (113), each of which will likely become increasingly significant given the advances of HTP top-down proteomics technologies (114) and the possibilities of a Human Proteoform Project (115).
Finally, Peter Uetz (Virginia Commonwealth University) described the use of yeast two-hybrid methods to detect genome-wide protein interaction networks (116) and how they can be used to provide functional clues regarding domains of unknown function (DUFs) (117). Uetz emphasized that essential DUFs can range from poorly to highly conserved (118) and that even well-studied housekeeping enzymes, such as those involved in glycolysis, can have regulatory or moonlighting roles through interaction networks that had not been appreciated until more recently (119).
The major point that emerged from all discussion groups was that most of the HTP data generated today are not easily mineable because they are not integrated or cross-referenced within most databases. It was emphasized that combining evolutionary associations with functional associations is a powerful way to discover new functions, but the latter are difficult to analyze as one often relies on supplemental excel files or deposited data with obsolete or unmappable identifiers. Ideally, BLAST-like engine(s) that can find homologs within various available HTP (e.g. expression data in model microbes or proteomics data, or ChIP-Seq data), such as the FitnessBrowser developed to analyze Tn-Seq data (120), would allow for the capture of all HTP data available for a given protein family. User-friendly tools for analyzing and comparing co-expression data from different organisms exist [see Table 1 of (121)] but vary greatly in the number of organisms covered and the usefulness of their respective outputs. Established databases like UniProt are always interested in ways to incorporate HTP data, but the lack of metadata and standards makes this objective difficult. In the rare cases where standards have been created and used by the community, such as the International Molecular Exchange standards for protein–protein interactions (122), then these data do get successfully integrated into databases and are much easier to mine. Such standards could allow easier capture of the essentiality data and phenotypes data that have now been gathered for many model organisms over the last 20 years (123, 124). The recent creation of the Global Biodata Coalition (https://globalbiodata.org/) has been a step in the right direction, providing stable funding for core databases that then could have more resources to work with for communities to create better data standards.
Another point of discussion that came up in several sessions in this meeting is the nonavailability of potential substrates for enzymatic or even phenotypic assays that greatly limit the power of HTP screens and require custom synthesis. Nor is it yet clear which HTP datatypes are optimal for understanding gene function, as different approaches (e.g. genetics, metabolomics and proteomics) are likely more or less informative for different protein classes. Benchmarking studies to ascertain the utility of these HTP data sets for understanding protein function in a few model organisms is required before the implementation of these approaches in additional organisms.
Synthesis, creating the roadmap for efficient and accurate functional annotation of the global proteome
Seán O’Donoghue (Garvan Institute), Chris Mungall (Berkeley National Laboratories) and Rich Roberts (New England Biolabs) gave the final three talks summarizing the main discussion points of the 1.5-day meeting (Figure 4) and proposing ideas for moving forwards.
Questions discussed during the five sessions.
A major recurrent theme throughout the meeting was the need for better communication between the different communities that work on functional annotations, mainly experimentalists, biocurators and computational biologists. Venues that bring these three communities together regularly do not exist. It was very clear that putting members of three groups in the same room (physical or virtual) for 2 days revealed that different languages and objectives had to be reconciled, but obvious cross-fertilization and problem-solving strategies also quickly emerged. Creating long-term sustainable collaborations across these three communities with different norms and schools of thought requires strategic and directed support—both before the collaborators come together and during their engagement. These could be spearheaded by societies working together. For example, the International Society for Biocuration could co-organize sessions at the general American Society for Microbiology (ASM) or Federation of American Societies for Experimental Biology (FASEB) meetings. Another possibility would be for the communities involved to apply for specific funding to enhance community building such as the NSF AccelNet (https://www.nsf.gov/pubs/2021/nsf21511/nsf21511.htm) or RCN https://www.nsf.gov/pubs/2017/nsf17594/nsf17594.htm) programs.
Another theme that emerged was that functional annotation must be democratized and become a global research community practice at the same level as deposition of raw genomics data in public repositories. Authors must become involved in this process. To make this possible, an infrastructure needs to be established to cement FAIR principles for protein functional annotation. The minimal/desired information about protein function needs to be defined, and the informatics infrastructure to allow annotation and curation of protein functional data at the time of paper submission needs to be implemented. Organizations already in place, such as Force 11 (https://force11.org), show that publishers and librarians are already primed to encourage and enforce FAIR functional annotation practices with publishing authors if the community were to just agree on a framework. Education of future scientists is also required as most biologists are unaware of how the data they generate get imported into the databases that they use. Several open-access training modules have already been created by EMBL/EBI focusing on biocuration (https://www.ebi.ac.uk/training/online/courses/biocuration-collection/) or data management; these should be integrated into biology graduate programs. Several successful efforts to involve students in annotation processes have already occurred, such as the HHMI SeaPhage Program, which has sequenced and annotated hundreds of phage genomes (https://seaphages.org), or the CACAO effort that uses undergraduates to check annotations (48). Students could also be involved in generating functional data like that which have been recently established by the SEA-GENES program funded by the Howard-Hughes Medical Institute where students screen several proteins for certain properties (https://www.hhmi.org/science-education/programs/science-education-alliance).
Building on his experience in creating GenBank and leading the Combrex effort (125), Rich Roberts strongly advocated for the creation of a consortium or umbrella organization (or a group within an existing one) that would take on the role of an advocate for articulating the importance of solving the functional annotation problem for all fields of life sciences. This consortium would take on the task of convincing politicians, private donors and/or companies of the need for specific funding. Importantly, the utility of measuring the economic impact of accurate and exhaustive protein functional annotation, an exercise recently performed by EBI (126), would go a long way to convince the different stakeholders.
A set of grand challenges that could be used to federate efforts and funding were identified. These include (i) integration and harmonization of all protein functional knowledge that is scattered across the literature and databases into a central resource or as common annotations in all databases. This might be a long-term objective, but the first step, which is improved interoperability between databases, is already in the works; (ii) predicting the context-dependent function of proteins in different organismal groups using an integrative model that takes into account sequence, structure, active sites, phylogenetic relationships, expression profile, subcellular localization, presence of substrates, etc.; (iii) identifying an incentive system for subject matter experts to provide annotation for databases. For example, a challenge could be correcting the poorly or wrongly annotated proteins in UniProt; (iv) creating high-quality annotations (integrating literature capture, models, expert curation, paralog flagging and confidence scores) for 100, 500 or 1000 representative genomes; (v) finding the genes for the 900 enzymes with EC numbers and no genes and (vi) identifying the function of ALL the genes in a few model organisms such as a yeast or E. coli. Funding and organizing communities to tackle a subset of these grand challenges could be a way to catalyze the required changes.
Conclusion
To conclude, mechanisms must be put in place to synergize, synthesize and democratize all aspects of the functional annotation ecosystem. Synergies need to be increased between communities (i.e. biocurators, computational biologists and experimentalists) to effectively transform expert experimental knowledge into high-quality, standardized functional annotations. HTP data sets must become easily mineable, so experimentalists who did not generate the data can readily use it to make functional hypotheses. Scientists working on non-model organisms need feedback from the model organism communities with successful stories of capturing all species-specific functional information to envision effective approaches for their biological domains of interest. Strategies already successfully applied for well-studied organisms (e.g. function curation projects and GO Phylogenetic Annotation) or specific protein superfamilies (e.g. SSNs for Radical SAM enzymes) could be scaled up and applied across the entire spectrum of protein diversity. The increased synergy between databases is critical to creating more highly connected resources of functional data (common IDs, centralized or federated repositories, consistent annotations, etc.). Finally, synergies between efforts to assess function prediction methods (e.g. Critical Assessment of protein Function Annotation (CAFA), Center for Critical Assessment of Genome Interpretation (CAGI), DREAM and BioCreative) would help leverage different approaches to address the various computational aspects of function prediction. The deficit of biocurators dedicated to recognizing, selecting, standardizing and integrating the vast amounts of experimental knowledge regarding specific proteins that remains untapped within the scientific literature needs to be recognized.
Supplementary data
Supplementary data are available at Database Online.
Acknowledgements
The conference attendees would like to thank Katherine MacWilkinson and her UF conference team for making the logistics of this hybrid meeting run so smoothly.
Funding
National Science Foundation (grant MCB-2129768) to V.d.C.-L; National Institutes of Health Intramural Research Program, National Library of Medicine (Z.L.).
Conflict of interest
None declared.
References
Author notes
Authors in alphabetical order.

{kind=link}
{kind=link}
{kind=link}
{kind=link}