





Abstract
The UniProt Knowledgebase (UniProtKB) acts as a central hub of protein knowledge by providing a unified view of protein sequence and functional information. Manual and automatic annotation procedures are used to add data directly to the database while extensive cross-referencing to more than 120 external databases provides access to additional relevant information in more specialized data collections. UniProtKB also integrates a range of data from other resources. All information is attributed to its original source, allowing users to trace the provenance of all data. The UniProt Consortium is committed to using and promoting common data exchange formats and technologies, and UniProtKB data is made freely available in a range of formats to facilitate integration with other databases.
Database URL:http://www.uniprot.org/
Introduction
The number of protein sequences in public sequence databases continues to grow exponentially as the number of completely sequenced genomes continues to increase. In addition, the amount of available information associated with these sequences is also increasing. This information is spread across a variety of biological data collections, necessitating a means of connecting all of this related but dispersed information so that users can seamlessly access it. Data integration plays an increasingly important role in bringing together the large amounts of diverse information spread across disparate resources and presenting a comprehensive overview of these data to the scientific community.
The UniProt Knowledgebase (UniProtKB) aims to act as a central hub of protein knowledge by providing a unified view of protein sequence and functional information. UniProtKB is produced by the UniProt Consortium which consists of groups from the European Bioinformatics Institute (EBI), the Swiss Institute of Bioinformatics (SIB) and the Protein Information Resource (PIR). The primary mission of the UniProt Consortium is to support biological research by maintaining a stable, comprehensive, fully classified, richly and accurately annotated protein sequence knowledgebase, with extensive cross-references and querying interfaces freely accessible to the scientific community.
UniProtKB consists of two sections, UniProtKB/Swiss-Prot and UniProtKB/TrEMBL. UniProtKB/Swiss-Prot is manually curated which means that the information in each entry is annotated and reviewed by a curator, while the records in UniProtKB/TrEMBL are automatically generated and are enriched with automatic annotation and classification. There are over 13.5 million entries in UniProtKB as of release 2011_01 of 11 January 2011 with 524 420 entries in UniProtKB/Swiss-Prot and 13 069 501 entries in UniProtKB/TrEMBL. UniProtKB is updated and distributed every 4 weeks and can be accessed online for searches or downloaded at www.uniprot.org.
Integrating sequence data
UniProtKB is a protein sequence database which aims to offer a complete collection of all publicly available sequences. To achieve this, it integrates sequences from a range of resources as summarized in Table 1. More than 99% of the sequences in UniProtKB are derived from translations of the coding regions in the International Nucleotide Sequence Database Collaboration (INSDC) which is composed of the European Nucleotide Archive (1), the DNA Data Bank of Japan (2) and GenBank (3). UniProtKB also accepts submissions of directly sequenced protein sequences through the web-based SPIN submission tool (4) which allows researchers to submit directly sequenced proteins and associated biological data. In addition, the published literature is searched on a monthly basis using literature databases such as CiteXplore (5) and UK PubMed Central (6) to identify papers reporting unsubmitted peptide sequence data for incorporation into the database. As part of an ongoing collaboration with PDBe (7), novel protein sequences are imported from the resource to ensure that all appropriate sequences in the worldwide Protein Data Bank (wwPDB) are represented in UniProtKB.
Sequence sources for UniProtKB
| Sequence sources | Data integrated into UniProtKB |
|---|---|
| DDBJ, ENA, GenBank | All protein sequences resulting from translations of annotated coding regions in the DDBJ, ENA and GenBank databases except for non-germline immunoglobulins and T-cell receptors, synthetic sequences, patent application sequences, small fragments of less than eight amino acids, and pseudogenes |
| Submissions | Directly sequenced protein sequences which have been submitted to UniProtKB |
| Literature | Directly sequenced protein sequences which have been published but which have not been submitted to a publicly available database |
| Protein Data Bank | Protein sequences for which a structure is available but for which there is no corresponding UniProtKB entry |
| Ensembl | Protein sequences resulting from gene predictions by the Ensembl group or manual curation from the Vega database for which there is no corresponding UniProtKB entry |
| RefSeq | Protein sequences resulting from gene predictions or manual curation by RefSeq for which there is no corresponding UniProtKB entry |
The International Protein Index (IPI) (Kersey et al.) (8) provides non-redundant complete proteome sets for a number of higher eukaryotic species and is used extensively by the proteomics community. It was launched in 2001 when information about proteomes was stored in diverse formats across many different databases. The situation has improved for many well-studied genomes and UniProtKB is now working in collaboration with Ensembl (9) and RefSeq (10) to provide complete protein sequence coverage of IPI organisms. A pipeline has been established to import novel human, mouse, rat, cow, dog, chicken and zebrafish sequences from Ensembl. UniProt will produce complete proteome sets for these species and, once this is completed, production of IPI will be discontinued. The pipeline will be expanded in the future to include all high-coverage Ensembl species. This pipeline facilitates the import of novel proteins based on gene predictions and allows the UniProt Consortium to draw on the expertise of the Ensembl group in this field. Ensembl also includes manually curated genes from the Vertebrate Genome Annotation (VEGA) database (11) which is particularly suited to annotation of splice variants which may be missed by automatic pipelines. The VEGA splice variants are manually identified on the basis of transcript evidence provided by cDNAs and/or ESTs. These additional splice variants are included in Ensembl and so are imported into UniProtKB and are used to supplement the set of splice variants identified by UniProtKB curators. Regular feedback is provided to Ensembl and VEGA if erroneous annotations from these sources are identified during the course of UniProtKB manual curation so that incorrect sequences may be updated or withdrawn. Incorrect predictions may be identified by comparison with available transcript and protein sequence data as well as with data from orthologous proteins in other species. This allows Ensembl to benefit from the manual curation expertise in the UniProt group. A similar pipeline to import novel sequences from RefSeq is currently being established.
This approach of importing and combining sequences from a range of sources ensures that UniProtKB provides a complete collection of protein sequences and also ensures consistency of proteome sets across sequence resources.
Annotation
UniProtKB adds value to each protein sequence record by including a wealth of information related to the role of the protein such as its function, structure, subcellular location, interactions with other proteins and domain composition, as well as a wide range of sequence features such as active sites and post-translational modifications. The information which is added directly to the database by the UniProt group comes from two main sources, manual curation and automatic annotation. Manual curation provides high-quality information for experimentally characterized proteins using data from the scientific literature as well as manual verification of results from sequence analysis programs. While manual curation is essential in providing accurate data, it is a time-consuming and labour intensive process which cannot keep up with the ever-increasing amounts of sequence data being generated. In addition, for many species, only the genome sequence has been determined with no functional experimental information available for the encoded proteins. To address these issues, automated methods have been developed which use information from known proteins to annotate uncharacterized proteins. Using both manual and automated curation approaches, as much information as possible is added to each UniProtKB record.
Manual curation
The manual curation process includes manual verification of each protein sequence as outlined below as well as a critical review of experimental data from literature and predicted data from a range of sequence analysis tools. Curators assimilate all of the information from these various sources, reconcile any conflicting results and compile the data into a concise but comprehensive report which provides a complete overview of the information available about a particular protein. The process consists of six major steps: (i) sequence curation, (ii) sequence analysis, (iii) literature curation, (iv) family-based curation, (v) evidence attribution and (vi) quality assurance and integration of completed entries. These steps ensure the quality and consistency of all manually curated data. The procedure is described in detail below and illustrated in Figure 1.
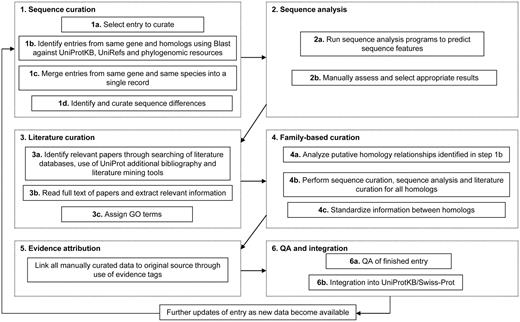
Flow diagram showing an outline of the UniProtKB manual curation process.
Sequence curation
Once a sequence has been selected for manual curation, BLAST (12) searches are run against UniProtKB to identify additional sequences from the same gene and to identify homologs. Sequences from the same gene and the same organism are compared and merged with all protein products encoded by one gene described in a single entry. This is done to reduce redundancy and ensures that users are provided with a comprehensive non-redundant collection of sequences where there is a single entry for each gene. A number of sequence alignment methods are used including T_Coffee (13), Muscle (14) and ClustalW (15) and these methods have been compared and reviewed in detail elsewhere (16, 17). Discrepancies between sequence reports are identified, and the underlying causes of the sequence differences such as alternative splicing, natural variations, frameshifts, incorrect initiation sites, incorrect exon boundaries and unidentified conflicts are documented. Comparison with homologous sequences is also used to identify additional sequence errors and their causes. These steps ensure that the sequence described for each protein in UniProtKB/Swiss-Prot is as complete and correct as possible and contribute to the accuracy and quality of further sequence analysis.
Sequence analysis
Sequences are analysed using a range of analysis tools for prediction of sequence features. The various tools have been integrated into an interactive sequence analysis platform that runs the programs simultaneously and displays the results in an interface that allows curators to review and select relevant results for inclusion. The predicted features include domains, repeats, transmembrane domains, secretory and organelle targeting sequences, coiled coils, regions of compositional bias, glycosylation sites, N-terminal myristoylation, GPI lipid anchor modification, and tyrosine sulfation. All predictions are manually reviewed and considered in the context of experimental data, and only relevant results are selected for integration. The full list of prediction methods used is described in Table 2.
Sequence analysis tools used during the UniProtKB manual curation process
Literature curation
Journal articles are the main source of experimental data. Relevant publications are identified by searching literature databases and using literature mining tools. The full text of each paper is read and relevant information is extracted for addition to the entry. The experimental data are critically assessed, summarized and compiled into a comprehensive report which provides a complete overview of the information available about a particular protein. The data are added in a highly structured and uniform manner using controlled vocabularies where possible to ensure consistency and to simplify data access. Annotation captured from the scientific literature includes protein and gene names, function, catalytic activity, co-factors, subcellular location, protein–protein interactions, patterns of expression, diseases associated with deficiencies in a protein, locations and roles of significant domains and sites, ion-, substrate- and co-factor-binding sites and catalytic residues as well as variant protein forms produced by natural genetic variation, RNA editing, alternative splicing, proteolytic processing and post-translational modification. A summary of the entry content is provided by the use of a number of keywords. UniProtKB keywords are a controlled vocabulary developed according to the need and content of UniProtKB/Swiss-Prot entries. They are used to index entries based on 10 categories: Biological process, Cellular component, Coding sequence diversity, Developmental stage, Disease, Domain, Ligand, Molecular function, Post-translation modification and Technical term. Each keyword is attributed manually to UniProtKB/Swiss-Prot entries and automatically to UniProtKB/TrEMBL entries according to specific annotation rules. The full list of keywords as well as definitions and mappings to corresponding Gene Ontology (GO) terms is available at http://www.uniprot.org/docs/keywlist. In addition, relevant GO terms (18, 19) are assigned based on experimental data from the literature.
Family-based curation
Reciprocal BLAST searches and phylogenetic resources such as Ensembl Compara (20) are used to identify putative homologs which are evaluated and curated. Annotation is standardized and propagated across homologous proteins to ensure data consistency. Functional information is propagated between orthologs and may also be transferred to paralogs if applicable.
Evidence attribution
All information added to an entry during the manual annotation process is linked to its original source so that users can trace the origin of each piece of information and evaluate it. The evidence attribution system and its use in both manual and automatic annotation procedures is described in more detail in a later section.
Quality assurance, integration and update
Each completed entry undergoes both automated and manual checks to ensure that it meets the required quality standards before integration into UniProtKB/Swiss-Prot. A quality control software program checks the syntax of each entry and also verifies a large number of biological rules such as the positions and relevance of amino acids cited in the entry, particularly regarding their roles as active sites or targets of post-translational modifications. Once an entry has passed the automated checks, it undergoes manual review to ensure that that all relevant sequences have been merged, that all relevant literature has been added, that the annotation has been added correctly, and that all relevant sequence analysis results have been included. This combination of automated and manual assessment of each entry ensures that the information content meets the required standards. Entries are updated on a regular basis as new data become available.
Automatic annotation
Records in UniProtKB lacking full manual curation are enhanced by the use of two complementary systems which aim to automatically annotate proteins with a high degree of accuracy. The UniRule system uses a set of rules which are manually created. The manual curation of the rules ensures the annotation quality of the system. In contrast, the Statistical Automatic Annotation System (SAAS) is a completely automated system where the rules are generated computationally using decision trees. The automatic generation of the rules ensures scalability of the system in the face of ever-increasing amounts of sequence data.
UniRule incorporates the HAMAP (21), RuleBase (22) and PIR (23) systems and applies annotation rules which are manually created and maintained by curators. Each rule specifies: (i) a number of annotations to be added by the rule based on information from experimentally characterized template entries and (ii) conditions which must be satisfied for the annotations to be applied. These conditions include family membership based on classification by InterPro (24), taxonomic restrictions, and the presence of particular sequence features. Predictions are evaluated against the content of manually annotated UniProtKB/Swiss-Prot entries as part of each UniProt release and rules that are inconsistent with current UniProtKB/Swiss-Prot annotation are reviewed and modified. This validation step ensures that only high-quality predictions are added and prevents propagation of potentially erroneous data.
SAAS generates automatic rules for functional annotation from UniProtKB/Swiss-Prot entries using the C4.5 decision tree algorithm (25). The algorithm was chosen because the derived rules are human-readable and short, and statistical evidence is given for each rule which can be used to order rules in terms of confidence (26). The algorithm determines the most concise rule for an annotation based on the criteria of sequence length, InterPro group membership and taxonomy. A data exclusion set is employed to ensure that only information suitable for computational annotation is predicted. The rules are generated as part of each release which ensures their evolution along with the UniProtKB with little or no manual intervention while also providing seed rules for exploitation in the UniRule system. SAAS includes a post-processing component for cross-validation against manually curated records to ensure the quality of the rules.
UniRule and SAAS together currently predict protein properties such as protein names, functions, catalytic activities, pathways, subcellular locations and sequence-specific information such as active sites for 34% of UniProtKB/TrEMBL entries. Numbers of predicted annotations from each system are provided in Table 3.
Numbers of predicted annotations from the UniProt automatic annotation systems for release 2011_01 of 11 January 2011
| Predicted annotations | Number of entries for which annotation is predicted | |
|---|---|---|
| SAAS | UniRule | |
| Protein names | N/A | 1 488 518 |
| Gene names | N/A | 583 214 |
| Comments | 1 455 030 | 2 929 410 |
| Keywords | 2 083 619 | 3 043 730 |
| Sequence features | N/A | 343 288 |
Cross-references
In addition to annotation added by both manual and automatic procedures, access is provided to information in more specialized resources through linking to relevant data by means of cross-references (27). Cross-references are provided to more than 120 different databases spanning a wide range of different resource types including nucleotide sequence resources, model organism databases and genomics and proteomics collections (Figure 2). The addition of a broad spectrum of cross-references ensures that UniProtKB acts as a central hub for biomolecular information by connecting to other resources which provide additional or complementary information.
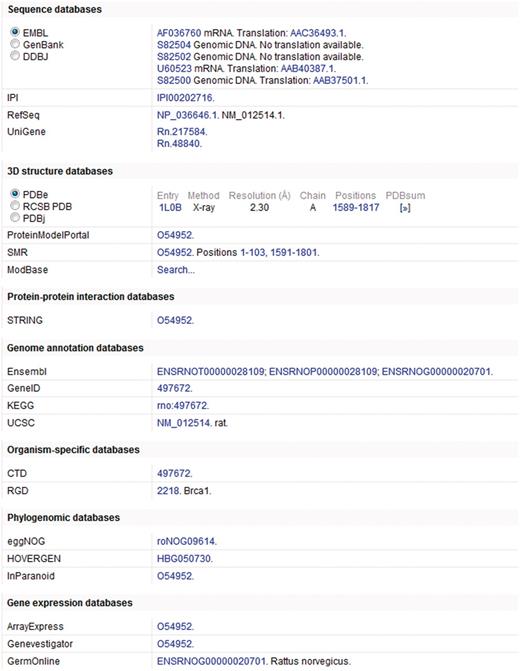
Cross-references in a UniProtKB entry. This figure shows a subset of the cross-references provided in UniProtKB entry O54952.
Cross-references are run every 4 weeks as part of each UniProt release to provide users with a complete set of regularly updated links. Establishing and maintaining these cross-references is the result of a collaborative effort with the scientific community, and contact with resource developers is maintained to ensure access to reliable and comprehensive data. In preparation for each cross-reference run, up-to-date mapping files are downloaded from each of the linked resources. These files are generated by the linked databases to provide a mapping of their entries to the corresponding UniProtKB entries and are used to generate the cross-references. Feedback is provided to the linked databases if incorrect mappings are identified during the curation process.
This ongoing contact and active collaboration with external resource providers ensures data quality and consistency. The incorporation of extensive cross-references allows UniProtKB to provide core data for a particular protein with easy access provided to complementary data in external resources. The full list of cross-referenced databases is provided at http://www.uniprot.org/docs/dbxref.
Annotation imported from other resources
As well as providing cross-references to external data collections, protocols have been established for importing data from selected resources into UniProtKB. These additional data supplement the information added during the manual and automatic annotation processes and ensure that UniProtKB provides a complete collection of integrated protein data. The following outlines some examples of the types of data which are imported from other data collections.
Nomenclature
UniProtKB imports gene names provided by official nomenclature committees as well as capturing other names used in the literature. Official gene names are imported directly from the appropriate species-specific database. This allows the standardization of nomenclature in line with recommended official names. Close collaborations exist with a number of species-specific resources such as the Human Gene Nomenclature Committee (HGNC) (28), the Mouse Genome Database (MGD) (29) and Flybase (30) and there is active communication between curators at UniProtKB and the various species-specific databases to ensure accurate data on both sides and to resolve any data inconsistencies. Collaboration takes place regarding issues such as ensuring linking of sequences to the correct gene, requesting new gene names or updates to existing gene names as required, and ensuring correct establishment of orthology relationships. New groups who wish to establish similar working relationships are encouraged to contact UniProt via help@uniprot.org.
Citations
As well as providing all citations which are used during the manual curation process, additional citations which have not been curated by UniProtKB are imported from a range of resources through close collaboration with the providers of other curated databases. This gives users access to additional publications absent from the UniProtKB record and allows them to better explore the available published literature for a particular protein. Citations are currently imported from 15 databases as shown in Table 4. These external sources contribute ∼475 000 unique PubMed citations which are not annotated in UniProtKB, covering ∼230 000 UniProtKB entries. The additional bibliography is directly linked from the protein entry view on the UniProt website where the additional citations can be accessed by clicking on the ‘Additional computationally mapped references’ link at the end of the References section.
Databases from which UniProtKB imports citations
| Database sources | Number of imported citations | Number of UniProtKB entries touched |
|---|---|---|
| BioCyc | 1780 | 1403 |
| dictyBase | 2530 | 2749 |
| Entrez Gene GeneRIF | 251 080 | 82 795 |
| FlyBase | 25 916 | 25 233 |
| GAD | 13698 | 24 042 |
| GeneDB_Spombe | 382 | 775 |
| MINT | 2521 | 26 181 |
| MGI | 110 796 | 54 016 |
| PDB | 16 455 | 15 575 |
| Reactome | 2574 | 3280 |
| RGD | 44 295 | 15 971 |
| SGD | 47 583 | 6316 |
| TAIR | 12 017 | 21409 |
| WormBase | 6747 | 8575 |
| ZFIN | 2987 | 6919 |
| Total | 475 490 | 230 991 |
Interactions
UniProtKB provides access to protein–protein interaction data in collaboration with the IntAct database of protein interactions (31) by importing a subset of high quality interactions from IntAct. The set of imported interactions is determined by the interaction detection method. A number of methods have been selected which are considered to produce accurate, reliable results and only interactions detected by these methods are imported. Examples of such methods are X-ray crystallography and surface plasmon resonance. Interactions identified by methods such as two-hybrid screening, which are known to produce a high number of false positive results, are imported only if they are confirmed by a second reliable method. IntAct is in the process of introducing a statistical scoring system which will be used in the future to determine which interactions are imported to UniProtKB. For each interaction, the gene name and accession number of the interacting protein are displayed along with the number of experiments in which the interaction has been observed (Figure 3). Specific information regarding the interaction is indicated in the ‘Notes’ column and each interaction is linked to the corresponding IntAct entry so that users can access more specific information for each interaction such as experimental details. This pipeline will be extended in the future to import interaction data from all members of the International Molecular Exchange (IMEx) Consortium. IMEx includes a number of interaction resources in addition to IntAct which perform non-overlapping curation of protein interaction data from distinct journals. Importing interaction data from all IMEx databases will broaden the coverage of interaction data in UniProtKB and ensure that it provides a complete non-redundant set of high-quality interaction data.
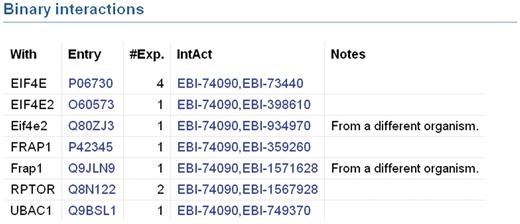
Binary protein–protein interactions in UniProtKB entry Q13541 which have been imported from IntAct. Each interaction is displayed on a separate line. The ‘With’ column contains the gene names of the interacting proteins. Accession numbers of interacting proteins are listed in the ‘Entry’ column. The ‘#Exp’ column provides the number of experiments in which an interaction has been observed. The ‘IntAct’ column contains the IntAct database accession numbers of the two interacting proteins. These are hyperlinked to provide users with access to the underlying data in the IntAct database. Specific information regarding the interaction may be present in the ‘Notes’ column.
GO terms
UniProtKB curators assign GO terms to all manually curated entries in the context of the Gene Ontology Annotation (GOA) (UniProtKB-GOA) project (32) which aims to provide high-quality GO annotations to proteins in UniProtKB. In addition to these manually assigned terms, GOA generates high-quality GO assignments using a number of electronic methods and also incorporates annotations from a range of other GO Consortium member databases. All of these GO terms are imported into the relevant UniProtKB entries along with details of the annotation source. This approach ensures maximum GO coverage while avoiding duplication of annotation across resources. UniProtKB-GOA currently provides GO annotations for 66% of UniProtKB entries (see Table 5 for current UniProtKB-GOA statistics).
Coverage of UniProtKB-GOA annotation
| Annotation source | Number of associations | Number of distinct UniProtKB proteins |
|---|---|---|
| Electronic annotations | 74 764 592 | 9 001 654 |
| Manual annotations by UniProt | 129 305 | 27 554 |
| Total manual annotations | 736 895 | 113 675 |
| Total GOA annotations | 75 501 487 | 9 015 498 |
The data are based on UniProtKB-GOA release 91 which was released on 12 January 2011 and was assembled using the publicly released data available in the source databases on 10 January 2011. A more detailed breakdown which is updated with each release is available at http://www.ebi.ac.uk/GOA/uniprot_release.html.
Evidence attribution
Each UniProtKB entry combines information from a wide range of sources including sequence data imported from DDBJ/ENA/GenBank, Ensembl and PDBe records, annotation imported from other databases, automatic annotation predictions and manually curated information based on experimental data from literature and results from sequence analysis programs. Because of this variety of data sources, it is vital that users are provided with a way of tracing the origin of each piece of information in an entry. The UniProt Consortium has developed a comprehensive evidence attribution system which attaches an evidence tag to each data item in a UniProtKB entry to indicate its source (Figure 4). Evidence tags are attached to data added during manual curation as described above and during automatic annotation as well as to data imported from external resources. The system provides users with a means of tracing the origin of each piece of information in an entry and evaluating it. Users can easily distinguish between experimental and predicted data and assess data reliability. In addition, the system facilitates the automatic correction and updating of data if the underlying source data changes while preserving manually curated information so that it is not overwritten by automatic procedures. The evidence attribution system is available in the XML version of UniProtKB and is also partially implemented in the entry view on the UniProt web site. Future plans include finer-grained tagging to allow a more detailed breakdown of source information, ongoing retrofitting of evidence tags to entries manually curated before the system was introduced, improved web site display, and modification of the system to make it compatible with the OBO Foundry Evidence Code Ontology which is already widely used by projects such as GO.
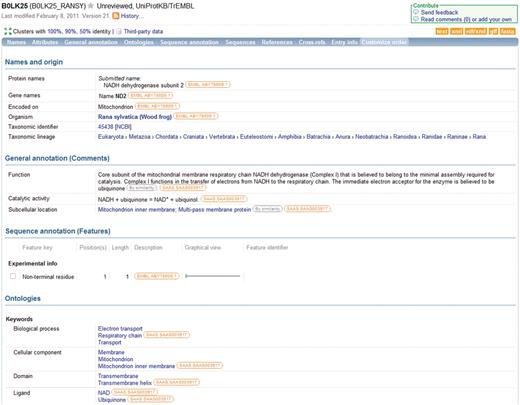
Information in a UniProtKB entry is linked to underlying data sources. The source of each data item is indicated and the source information is hyperlinked to allow users to access the original data source directly.
Use of Distributed Annotation System in data integration
UniProtKB makes use of the Distributed Annotation System (DAS) (33) to incorporate and display external data from multiple sources. DAS is a system for sharing and visualizing biological information using data provided by sources that are distributed around the world and remain under the control of the original provider. The system has been adopted by many data providers in the fields of genomics and proteomics. Data distribution is performed by DAS servers and is separated from visualization which is performed by DAS clients. The client-server architecture allows a single client to integrate information from multiple servers, collate the information, and display it to the user in a single view with little coordination needed among the various information providers.
The UniProt DAS server (34), which is available at http://www.ebi.ac.uk/das-srv/uniprot/das, provides access to sequences and annotation from both UniProtKB and the UniProt Archive (35), a comprehensive and non-redundant database that contains all of the protein sequences from the main publicly available protein sequence databases. Research groups can provide and view their own data in the context of UniProtKB annotations and UniParc cross-references through the use of a suitable DAS client. The server also gives access to Gene Ontology annotation of UniProtKB proteins and to theoretical tryptic digests of protein sequences in UniProtKB.
In addition, the UniProt web site provides access to the Dasty2 web client (36) for visualizing protein sequence feature information from more than 40 DAS servers. Dasty2 integrates and merges sequence annotations from multiple sources and also displays sequence details and other information such as publications and protein structures when available. The data are provided in a unified interactive graphical view which facilitates rapid searches to find, share and compare annotations for a protein of interest. The client is accessible from each UniProtKB entry on the UniProt web site through the ‘Third-party data’ link which can be found at the top of each entry page.
Accessing UniProtKB data
All UniProtKB data is freely available from the UniProt web site (37) at www.uniprot.org. The web site provides tools for querying and analysing the data as well as a wide range of documentation, and supports full text and field-based text searches, sequence similarity searching, multiple sequence alignments, batch retrieval and database identifier mapping.
A Google-like full-text search is provided as the main entry point. In addition, searches can be built iteratively using the query builder (Figure 5) or can be entered manually in the query field which can be faster and more powerful (see http://www.uniprot.org/help/text-search for details of query syntax). Viewing of result sets and individual entries is configurable. At the level of result sets, results are returned in a table which users can customise with respect to the types and order of columns displayed, and the number of rows displayed per page. At the level of an individual entry, users can customize the order in which entry sections are displayed. User customizations are saved and all queries can be bookmarked so that they can be repeated on new releases of the data.
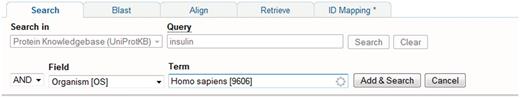
Using the query builder on the UniProt website to refine a search. An initial query for insulin is further refined using the query builder to include a taxonomic restriction.
In addition to text searches, sequence similarity searches are a commonly used way to search UniProtKB, and BLAST is provided for this purpose. Searches can be run against multiple databases, it is possible to restrict the search to particular taxonomic groups and the results display can be customized. For running multiple sequence alignments, ClustalW is provided. A batch retrieval tool allows users to enter a set of UniProt identifiers and retrieve the corresponding entries. To allow users to map lists of gene or protein identifiers to UniProtKB proteins, an identifier mapping tool is provided (Figure 6). The tool takes a list of UniProt identifiers as input and maps them to identifiers in a database referenced from UniProt or vice versa.
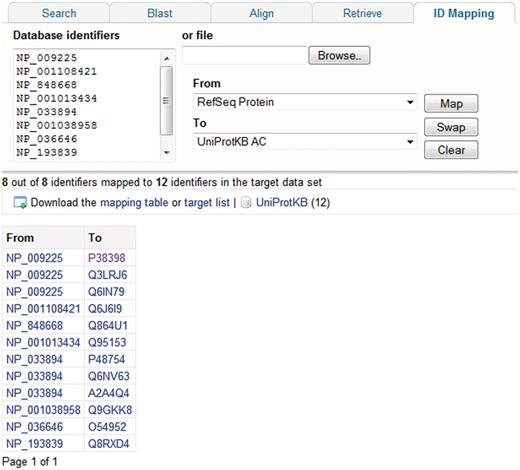
Mapping database identifiers using the identifier mapping tool on the UniProt website. The identifier mapping tool allows mapping of UniProt identifiers to identifiers in a database referenced from UniProt or vice versa. Here, a set of RefSeq identifiers are mapped to the corresponding UniProtKB entries.
UniProtKB data is released every 4 weeks as part of each UniProt release and is provided in a range of formats, depending on the chosen data set, to aid seamless exchange with other resources. The formats provided include plain text, XML, RDF and GFF for data files, and FASTA format for sequence files. Releases are versioned using the format YYYY_XX where YYYY is the calendar year and XX is a two-digit number that is incremented for each release of a given year, e.g. 2011_01, 2011_02, etc. Previous releases are archived on the UniProt FTP site for at least 2 years.
Programmatic access to data and search results is provided via simple HTTP (REST) requests. This facilitates the development of applications using UniProtKB data and supports commonly used data formats. Full details and code examples are available on the UniProt web site at http://www.uniprot.org/faq/28. In addition, a Java application programming interface, the UniProtJAPI (38), has been developed to provide remote access for Java applications processing UniProt data, and facilitates the integration of UniProt data into Java-based software applications. The library supports queries and similarity searches that return UniProtKB entries in the form of Java objects.
Integrated data querying
While the UniProt web site provides a query interface which allows the searching of all UniProtKB data, biologists often need to perform complex queries across a variety of databases. BioMart (39) is an open-source data management system that allows for integrated querying of biological data resources regardless of their geographical locations. It was developed to enable scientists to perform advanced querying of multiple biological data sources through a single web interface. The BioMart model eliminates the need to aggregate and manage the data in a central location which means that individual data providers remain responsible for updates and release cycles, and it also removes the need for users to become familiar with the query interfaces of multiple individual resources. The UniProt BioMart (http://www.ebi.ac.uk/uniprot/biomart/martview) allows users to perform complex queries across UniProtKB, InterPro, Ensembl and PRIDE (40) which are not possible to perform on the UniProt web site. Examples of queries which can be performed are ‘Give me the DNA sequence in Ensembl for a given protein sequence in UniProt’ or ‘Give me all proteins from UniProtKB that have been reported as identified in PRIDE and which are referenced in UniProtKB to a particular OMIM entry’.
Future plans
Manual curation will continue to provide high-quality UniProtKB data, ensuring that users have access to accurate and consistently annotated experimental information coupled with manually verified sequence analysis predictions. In addition, the automatic annotation systems will be improved and expanded to increase the depth and breadth of predicted data while ensuring the continued quality of the predicted annotations. Existing cross-references will continue to be maintained and regularly updated with each release and new cross-references will be added to the collection as appropriate.
The UniProt Consortium will continue to explore additional high-quality data import sources and a number of new data sets will be introduced in the near future. Variant data will be imported from Ensembl to complement the literature-based variant data in UniProtKB which, in turn, will be provided to Ensembl to supplement their variant set.
Building on the close collaboration which already exists between UniProtKB and the wwPDB, data related to interactions of proteins and small molecules will be imported from the PDBeMotif database (41). This will include positional information for binding sites along with associated literature citations. Given the broad range of chemical entities contained in the PDB, only a manually chosen subset of unambiguously biologically relevant molecules will be included. UniProtKB will also extend its collaborations with proteomics resources such as PRIDE to incorporate mass spectrometry-derived data sets.
Conclusions
Data integration is essential to ensure that users have access to a unified view of the growing body of biological information which is spread across multiple resources. The UniProt approach to data integration ensures that information is captured in the most appropriate resource for subsequent integration with other databases and also ensures maximum curation efficiency by preventing duplication of efforts across multiple resources.
Funding
National Institutes of Health (2U01HG02712-04); Additional support for EBI’s involvement in UniProt comes from the European Commission contract SLING grant (226073); National Institutes of Health (2P41HG02273-07); UniProtKB/Swiss-Prot activities at the SIB are supported in addition by the Swiss Federal Government through the Federal Office of Education and Science and by the European Commission contracts GEN2PHEN (200754); MICROME (222886-2); and SLING (226073);; PIR activities are also supported by the National Institutes of Health (5R01GM080646-04, 3R01GM080646-04S2, 1G08LM010720-01 and 3P20RR016472-09S2); and the National Science Foundation (DBI-0850319). Funding for open access charge: National Institutes of Health (1U41HG006104-01).
Conflict of interest. None declared.
Acknowledgements
UniProt has been prepared by:
Rolf Apweiler, Maria Jesus Martin, Claire O’Donovan, Michele Magrane, Yasmin Alam-Faruque, Ricardo Antunes, Benoit Bely, Mark Bingley, David Binns, Lawrence Bower, Paul Browne, Wei Mun Chan, Emily Dimmer, Ruth Eberhardt, Francesco Fazzini, Alexander Fedotov, John Garavelli, Leyla Garcia Castro, Rachael Huntley, Julius Jacobsen, Michael Kleen, Kati Laiho, Duncan Legge, Wudong Liu, Jie Luo, Sandra Orchard, Samuel Patient, Klemens Pichler, Diego Poggioli, Nikolas Pontikos, Manuela Pruess, Steven Rosanoff, Tony Sawford, Harminder Sehra, Edward Turner, Matt Corbett, Mike Donnelly and Pieter van Rensburg at the European Bioinformatics Institute;
Ioannis Xenarios, Lydie Bougueleret, Andrea Auchincloss, Ghislaine Argoud-Puy, Kristian Axelsen, Amos Bairoch, Delphine Baratin, Marie-Claude Blatter, Brigitte Boeckmann, Jerven Bolleman, Laurent Bollondi, Emmanuel Boutet, Silvia Braconi Quintaje, Lionel Breuza, Alan Bridge, Edouard deCastro, Elisabeth Coudert, Isabelle Cusin, Mikael Doche, Dolnide Dornevil, Severine Duvaud, Anne Estreicher, Livia Famiglietti, Marc Feuermann, Sebastien Gehant, Serenella Ferro, Elisabeth Gasteiger, Alain Gateau, Vivienne Gerritsen, Arnaud Gos, Nadine Gruaz-Gumowski, Ursula Hinz, Chantal Hulo, Nicolas Hulo, Janet James, Silvia Jimenez, Florence Jungo, Thomas Kappler, Guillaume Keller, Vicente Lara, Philippe Lemercier, Damien Lieberherr, Xavier Martin, Patrick Masson, Madelaine Moinat, Anne Morgat, Salvo Paesano, Ivo Pedruzzi, Sandrine Pilbout, Sylvain Poux, Monica Pozzato, Nicole Redaschi, Catherine Rivoire, Bernd Roechert, Michel Schneider, Christian Sigrist, Karin Sonesson, Sylvie Staehli, Eleanor Stanley, Andre Stutz, Shyamala Sundaram, Michael Tognolli, Laure Verbregue, Anne-Lise Veuthey at the Swiss Institute of Bioinformatics;
Cathy H. Wu, Cecilia N. Arighi, Leslie Arminski, Winona C. Barker, Chuming Chen, Yongxing Chen, Pratibha Dubey, Hongzhan Huang, Raja Mazumder, Peter McGarvey, Darren A. Natale, Thanemozhi G. Natarajan, Jules Nchoutmboube, Natalia V. Roberts, Baris E. Suzek, Uzoamaka Ugochukwu, C. R. Vinayaka, Qinghua Wang, Yuqi Wang, Lai-Su Yeh and Jian Zhang at the Protein Information Resource.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}