





Abstract
Annotation using Gene Ontology (GO) terms is one of the most important ways in which biological information about specific gene products can be expressed in a searchable, computable form that may be compared across genomes and organisms. Because literature-based GO annotations are often used to propagate functional predictions between related proteins, their accuracy is critically important. We present a strategy that employs a comparison of literature-based annotations with computational predictions to identify and prioritize genes whose annotations need review. Using this method, we show that comparison of manually assigned ‘unknown’ annotations in the Saccharomyces Genome Database (SGD) with InterPro-based predictions can identify annotations that need to be updated. A survey of literature-based annotations and computational predictions made by the Gene Ontology Annotation (GOA) project at the European Bioinformatics Institute (EBI) across several other databases shows that this comparison strategy could be used to maintain and improve the quality of GO annotations for other organisms besides yeast. The survey also shows that although GOA-assigned predictions are the most comprehensive source of functional information for many genomes, a large proportion of genes in a variety of different organisms entirely lack these predictions but do have manual annotations. This underscores the critical need for manually performed, literature-based curation to provide functional information about genes that are outside the scope of widely used computational methods. Thus, the combination of manual and computational methods is essential to provide the most accurate and complete functional annotation of a genome.
Database URL:http://www.yeastgenome.org
Introduction
Generating gene ontology annotations
Since its inception in 1999, Gene Ontology (GO) has become the standard for functional annotation, and is used by all model organism databases as well as by genome projects for less-characterized organisms (1, 2). The GO is a controlled, structured vocabulary for annotating gene products according to the molecular functions that they perform, the biological processes in which they participate, and the cellular components in which they reside (3, 4). The core of a GO annotation is comprised of a GO term representing a function, process or location, and an evidence code indicating the basis for the assignment, linked to a gene product and the reference for the observation. The GO annotations that are present in model organism databases can be broadly divided into two categories: literature-based annotations, based on scientific publications, and computational predictions, generated by automated methods.
Literature-based annotations are derived from published information by trained, PhD-level scientific curators. The process begins with the identification and prioritization of the relevant literature for curation. This presents particular challenges for each database, depending on the size of the research community and rate of publication for that organism. Even the first step, identifying the species and genes studied, may require a significant effort (5, 6). Once a body of literature relevant to specific genes or proteins of an organism has been identified, it is a further challenge to select and prioritize papers containing information that can be captured as GO annotations. Studies are underway to apply automated methods to this process (7–11), but it is still largely a judgment call by a curator who has skimmed the abstracts or full text of the papers. After papers are selected for GO curation, creating the annotations requires the scientific expertise of the trained curator, who has broad knowledge of the biology of the organism in question, is familiar with the GO content and structure, and is experienced in the standard curation procedures for GO annotation such as the use of evidence codes, qualifiers and other details. The aim is not simply to apply every possible GO term to a gene product, but rather to annotate with the most current and direct information, in the context of all available knowledge. For example, at the Saccharomyces Genome Database (SGD), if the only experimental result for a gene were the mutant phenotype of abnormal bud site selection, we would annotate using a Biological Process term for ‘cellular bud site selection’ (GO:0000282). However, if a different gene had the same phenotype but was also known to be involved in a more specific process, such as ‘mRNA export from nucleus’ (GO:0006406), it would not receive the GO annotation to ‘cellular bud site selection’, since the phenotype represents an indirect effect of the defect in the primary process. The mutant phenotype would, however, be captured for all relevant genes using SGD’s mutant phenotype curation system (12). Thus, the GO annotations reflect the processes in which the current state of the literature indicates that each gene product is directly and specifically involved, while the mutant phenotype annotations reflect the comprehensive set of phenotypes observed, whether directly or indirectly related to the role of the gene product (12).
It is obvious that manual curation by experienced curators is very valuable, and it is generally considered the ‘gold standard’ for annotation. The annotations are chosen carefully within the biological context of all available knowledge about a gene product, and represent the best possible summary of the most relevant information about it (13, 14). On the other hand, literature-based curation is very time- and resource-intensive. In order to ensure that it is of the best possible quality, a comprehensive set of annotations must be generated by curating all of the literature specific to the gene products of an organism, and new literature must be curated as it is published (15). In addition to continually generating new annotations, existing annotations must also be reviewed and updated regularly. Annotations that are accurate at the time of their creation may become ‘stale’ for various reasons: if the GO structure changes, for example if a new, more granular term is added that is appropriate to the gene product; or if new characterization of a gene product means that a previously annotated role is seen to be indirect or is proven to be untrue. Finally, since human variability and error cannot be avoided, there is always the possibility that literature-based annotations are incomplete or inconsistently assigned.
In contrast, computational predictions do not require experimental work on the organism to which they are applied, other than a genome sequence. They can be assigned very rapidly, with minimal human effort, and the annotation parameters are uniform across all gene products. It is also straightforward to re-run the computation on a regular basis, in order to keep up with improvements to the computational methods, changes to the genome sequence annotation or changes to GO. However, there are limitations to computational predictions, in that their scope may not be as broad as that of literature-based annotations; they often use very general GO terms; and different methods may have different inherent biases.
GO annotations at SGD
With its complete genomic sequence available since 1996, curation of functional data by SGD since 1994, and experimental characterization for 84% of protein coding genes currently available, S. cerevisiae is a rich source of functional annotation for comparative studies (16–18). SGD curates and integrates a myriad of datatypes for the yeast community, including the genomic sequence; gene names, synonyms and descriptions; mutant phenotypes; genetic and physical interactions; and expression data. Although GO annotations are just one aspect of curation, they capture a very large proportion of the functional information that is available. Including both literature-based annotations and computational predictions, the SGD gene association file (GAF, http://downloads.yeastgenome.org/literature_curation/) from November 2010 contains approximately 64 000 GO annotations (counted as unique pairs of a gene and GO term) for more than 6300 gene products. On average, 70 new research papers are loaded each week, and SGD curators add an average of 80 GO annotations for 24 gene products. An additional body of about 1200 papers has been marked by curators as possibly containing GO-curatable information, and awaits further curation. Because of the magnitude of the existing and ongoing GO curation, combined with finite resources, it is imperative to develop efficient ways of identifying and prioritizing annotations for review and updating in order to maintain the high quality upon which many projects depend.
In addition to literature-based GO annotations, SGD contains a large set of predictions based on computational analyses performed outside of SGD. The majority of computational predictions available at SGD are performed by the Gene Ontology Annotation (GOA) project, which is part of the UniProt group at the European Bioinformatics Institute. Since the methods used by the GOA project are applied consistently to all proteins in all species, the GOA predictions provide a dataset that is comparable across multiple genomes. SGD’s GAF includes all computational predictions made by the GOA project for S. cerevisiae, comprised of results from four methods that map several different types of information to GO terms (19). InterPro entries, which represent conserved protein sequence patterns such as domains, motifs, active sites or protein family signatures (20, 21) are mapped to GO terms that represent the role of proteins bearing that particular pattern. Swiss-Prot Keyword (SPKW) analysis assigns GO annotations based on mappings to keywords that are found in UniProtKB/Swiss-Prot entries (http://www.geneontology.org/external2go/spkw2go): for example, a protein whose entry contains ‘antiport’ would be assigned the GO Molecular Function term ‘antiporter activity’. E.C. to GO mappings (http://www.geneontology.org/external2go/ec2go) indicate the GO terms that correspond to the Enzyme Commission numbers used to classify enzymatic activities. The subcellular location terms found in UniProtKB/Swiss-Prot entries (SPSL) are also mapped to GO Cellular Component terms (http://www.geneontology.org/external2go/spsl2go). In addition to these predictions from the GOA project, SGD also contains computational predictions generated by two sophisticated algorithms. The bioPIXIE method, developed by the Troyanskaya group, integrates more than 700 diverse datasets (for example, genomic expression, protein–protein interaction, protein localization) to predict the biological processes in which gene products participate (22–24). The YeastFunc method, from the Roth group, also incorporates biological relationships inferred from various large-scale datasets, as well as sequence-based predictions, to generate GO annotations in all three aspects (25). The GO annotations in SGD that are assigned via all of these computational methods carry the IEA (Inferred from Electronic Annotation) or RCA (Reviewed Computational Analysis) evidence codes.
Here, we present a strategy for comparing GO annotations generated by literature-based and computational approaches. We apply this strategy to one subset of annotations in SGD, and explore the feasibility of extending the strategy to additional sets of annotations and expanding it to organisms beyond yeast.
Results and discussion
Leveraging InterPro predictions to update ‘unknown’ annotations
The GO annotations generated by SGD are used for many purposes: for basic research on yeast; for comparative genomics, as a source of functional predictions for other organisms; and as a training set for the development of computational prediction algorithms (18). Because of their importance to so many different endeavors and their propagation beyond SGD, it is critical that they are as accurate and as comprehensive as possible. We decided to explore whether our large set of computational predictions could be leveraged to find inaccuracies or omissions in literature-based GO annotations, allowing us to identify and prioritize genes for review and updating. For an initial feasibility study, we chose to analyze the InterPro signature-based predictions, reasoning that since this method is very widely used for a variety of organisms, our conclusions might be applicable to other databases in addition to SGD.
To identify annotations that needed to be reviewed and updated, all literature-based annotations were compared with all InterPro-based computational predictions to generate pairs of literature-based and computational annotations when both types of annotation existed for a given gene and GO aspect (Function, Process or Component). These annotation pairs could be classified into different groups according to the relationship between the two GO terms. For example, a pair containing a literature-based annotation and a computational prediction may have the same GO term, may have two terms that are in the same lineage of the GO structure or may have terms in different GO lineages. Some of these relationships might highlight areas where literature-based annotations were incomplete, incorrect or not as granular as possible.
As a first step in assessing the utility of such a comparison, we focused on a specific subset of literature-based annotation and computational prediction pairs. Specifically, we asked whether computational predictions could help in updating genes to which curators were previously unable to assign a literature-based GO annotation. When the gene product-specific literature for a species has been curated or searched comprehensively, and a curator has found no published information that would allow assignment of a GO annotation for a particular gene product and GO aspect, this fact is captured by assignment of an ‘unknown’ annotation. In practice, these annotations are created by assigning the root terms in each aspect, which are the broadest terms that exist: ‘molecular_function’ (GO:0003674), ‘biological_process’ (GO:0008150) and ‘cellular_component’ (GO:0005557). For convenience, we will refer to these annotations as ‘unknown’. Annotating with these terms allows the distinction to be made between the absence of published results and the absence of an annotation. The presence of an ‘unknown’ annotation indicates a curator has determined that there was no published information useful in assigning a GO annotation on the date of review. The complete absence of an annotation means that a curator has not yet performed a review of the literature (2, 18).
We were interested to determine whether comparison with computational predictions would identify ‘unknown’ annotations that could now be updated to more informative annotations—either because new experimental information had been published, or because such information existed previously but had been overlooked. SGD curation and loading of InterPro predictions from the GOA project are both constantly ongoing, so in order to work with a static dataset, we analyzed the complete sets of literature-based and computational GO annotations from SGD, as well as the GO ontology file, dating from October 2009.
Considering all three GO aspects together, 4129 of the 31 977 literature-based annotations in this October 2009 set were ‘unknowns’. A corresponding InterPro-based prediction existed for 608 of the ‘unknown’ annotations. We reviewed 67 of these (including a representative set from each aspect, comprising 10% of the annotations in that aspect), to assess whether we could update the literature-based annotations based on the computational predictions. The results of this analysis are presented in Table 1.
Review of curator-assigned ‘unknown’ annotations (to the root terms) for which a corresponding InterPro prediction was available
| Annotations | Genes | |
|---|---|---|
| Total number of ‘unknowns’ | 4129 | 2318 |
| ‘Unknowns’ with predictions | 608 | 477 |
| ‘Unknowns’ with predictions reviewed | 67 | 57 |
| ‘Unknowns’ with predictions updated | 16 | 16 |
All data are from October 2009.
For the majority of ‘unknown’ annotations reviewed (51 annotations, 76%), the literature review uncovered no additional evidence that allowed us to update them; for the remaining 24% (16 annotations), we were able to make updates. In most of the cases where an update was possible, we were able to replace the ‘unknown’ with a literature-based annotation to the same GO term that was used for the prediction, or with a term in the same branch of the ontology. In a few cases, we could not apply the particular GO term suggested by the InterPro prediction, but during the course of reviewing the literature we found evidence that allowed us to add or update GO annotations for that gene. In both of these instances, we consider that the InterPro prediction was helpful because if those annotations had not been flagged for review by the presence of a prediction, we would not have prioritized those genes for review.
In order to assess whether the InterPro predictions were significantly helpful in identifying manual annotations that could be updated, we compared these results to those obtained by analysis of a comparable, ‘control’ set. This control set was randomly chosen from the full set of ‘unknown’ annotations with no corresponding InterPro predictions. The same number of annotations in each GO aspect was reviewed in the control set as the experimental set. In each case, we reviewed the literature to see whether there was any available evidence that would allow us to replace the ‘unknown’ with a literature-based annotation. We found published evidence that would allow us to update 7 out of the 67 annotations (10.4%). This is a significant difference from the previously reviewed set (16 updates out of 67 annotations reviewed; P < 0.001, chi-squared test), indicating that the presence of an InterPro prediction corresponding to a manual ‘unknown’ annotation identifies a set of annotations whose review may be productive.
Despite the fact that the set of manual ‘unknown’ annotations with corresponding InterPro predictions was enriched for annotations that could be updated, the enrichment was relatively small relative to the effort required to make the updates. To update the 16 ‘unknown’ annotations with InterPro predictions, we reviewed the literature for all 57 genes in the sample set (634 publications in total) in order to determine whether there was any experimental evidence to support the predictions. On average, 40 papers were reviewed in order to make each annotation change. Extrapolating from the sample set, we would estimate that application of this review process to our total set of 608 manual ‘unknown’ annotations that have corresponding InterPro predictions would allow us to update 145 of the annotations. These updates would require reviewing the total of 2987 papers that are associated with the 477 genes in this set. While these updates are certainly valuable for the particular genes that they affect, they are relatively insignificant when considered in the context of all SGD annotations (Figure 1). Thus, although the method was effective for the ‘unknown’ annotations that were updated, it was not very efficient in terms of curation effort. However, we explore below the possibility that comparison of the much larger set of non-‘unknown’ literature-based annotations to computational predictions might be more productive.
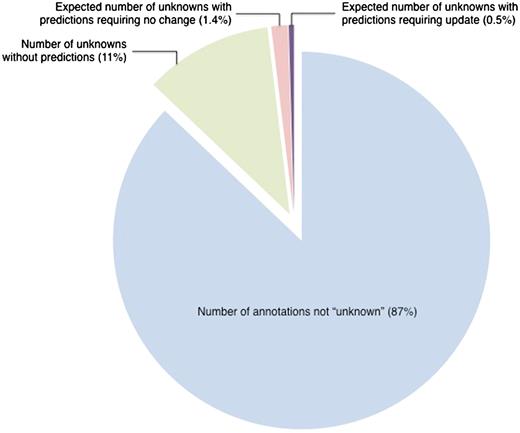
Estimated number of ‘unknown’ annotations that could be updated by comparison to InterPro-based computational predictions. Out of 31 977 literature-based GO annotations, 4129 annotations, representing 13% of all annotations, are to the root terms ‘molecular_function’, ‘biological_process’, ‘cellular_component’ (referred to as ‘unknown’). Based on a review of a representative set, only 145 ‘unknown’ annotations are projected to need review. This represents 0.5% of the entire set of annotations. All data are from October 2009.
Assessing the scope of computational predictions
As a further step in assessing the feasibility of this method, we investigated the scope of computational predictions, since any strategy comparing literature-based annotations with computational predictions can only be effective if both are available for the comparison. We determined the percentage of the genome covered by both kinds of annotation, starting with S. cerevisiae and then extending the analysis to several different organisms. All data for this analysis were downloaded from the respective databases (see below) in November 2010.
SGD’s gene association file (GAF) contains GO annotations for 6357 genes, including RNA-coding as well as protein-coding genes. We first considered InterPro signature-based computational predictions, and determined that there are 2803 genes in SGD (44%) that are not covered by these predictions; that is, they have literature-based annotations only (Figure 2). The set of genes that lack predictions not only includes the RNA-coding genes, as expected for a protein sequence-based method, but also a substantial fraction of protein-coding genes. To see whether the additional predictions from the GOA project would add to the coverage, we expanded the analysis to include predictions derived from E.C. numbers, Swiss-Prot keywords and Swiss-Prot Subcellular Locations. When these methods are included, there are still 1025 S. cerevisiae genes lacking computational predictions.
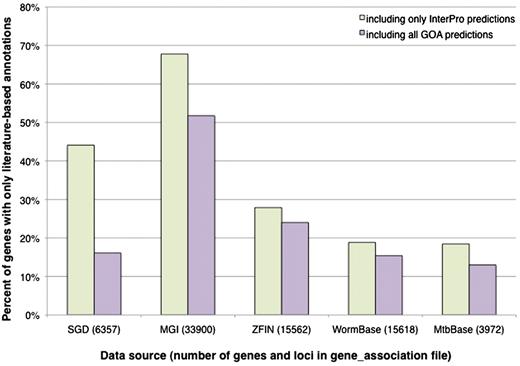
Percentage of genes with literature-based annotations and no computational predictions. Genes from each data source were determined to have a literature-based annotation, a computational prediction from an InterPro signature, or a computational prediction from any of the methods used by the GOA project. All of the computational predictions were performed by the GOA project, using consistent methods and parameters, and are incorporated in their entirety by the various model organism databases. The graph displays the proportion of genes in each organism that have only literature-based annotations when compared to InterPro-based predictions only, or when compared with all predictions made by the GOA project (including those based on InterPro signatures). All data, including the total number of genes and loci listed for each data source, are based on computational annotations (identified as a GO annotation with an IEA evidence code) found in gene association files downloaded in November 2010.
To determine whether this trend is unique to SGD, we examined the coverage of literature-based annotations and computational predictions available for genes in the model organisms Caenorhabditis elegans (WormBase, http://www.wormbase.org/), Danio rerio (ZFIN, http://zfin.org/) and Mus musculus (MGI, http://www.informatics.jax.org/), and for the pathogen Mycobacterium tuberculosis (TBDB, http://www.tbdb.org/; MTBbase, http://www.ark.in-berlin.de/Site/MTBbase.html). These database groups were selected because they include computational predictions from the GOA project in their entirety in their GAF. For each organism, we obtained the GAF from the respective database, determined the total number of genes represented in it, and determined the number of genes for which there are literature-based GO annotations but no computational predictions made by the GOA project (as identified by an IEA evidence code and appropriate reference in the GAF). We performed the comparison of genes with literature-based annotations to genes with computational predictions, as described above for S. cerevisiae, considering either InterPro-based predictions alone or expanding the analysis to include InterPro and any additional prediction methods applied to that organism by the GOA project.
The data for all organisms, presented in Figure 2, show the proportion of genes having literature-based annotations only, and lacking computational predictions. It is evident that S. cerevisiae is not unique. In the model organism databases, SGD and WormBase contain roughly the same proportion of genes with literature-based annotations but lacking computational predictions (∼16%), while the proportion is higher in ZFIN (24%), and in MGI, over 50% of mouse genes with literature-based annotations lack computational predictions. As an example of a less annotated organism for which computational predictions may be particularly important, M. tuberculosis has a relatively recent genome sequence and a small, privately funded literature-based GO annotation effort that has not yet reached comprehensive coverage of the literature (MTBbase, http://www.ark.in-berlin.de/Site/MTBbase.html). Still, ∼13% of M. tuberculosis genes have literature-based annotations but no computational predictions made by the GOA project. It is apparent that for all these organisms, there is a subset of genes that is not covered by any of the GOA prediction methods. This is consistent with the statistic that the GOA database (including both computational predictions and literature-based annotations) did not include any annotations for ∼31% of the entries in UniProtKB in 2005 (7).
We draw two conclusions from this feasibility study. First, a substantial proportion of genes in each genome do have literature-based annotations as well as computational predictions. Therefore, these genes would be able to benefit from a quality control method that requires the existence of a computational prediction with which to compare a literature-based annotation. In the future, we will focus on whether computational predictions made by the GOA project can be helpful in identifying and prioritizing S. cerevisiae annotations that need to be reviewed and updated. Second, it is apparent that a proportion of genes in each genome is outside the scope of computational predictions made by the GOA project, and has only literature-based GO annotations. Computational predictions made by the GOA project represent the most comprehensive effort to provide functional information for all proteins from all species. Millions of proteins in UniProtKB that are experimentally uncharacterized would have no GO annotations if it were not for GOA computational predictions (19). However, manual literature-based curation efforts are still needed to provide a complete functional description of a genome.
Summary and conclusions
In this era of increased data generation coupled with decreased funding for curation efforts (5, 26), it is critical to develop innovative and efficient strategies for prioritizing annotations for review, in order to maintain the extremely high quality of literature-based annotation sets. We report here the first steps in establishing a procedure for leveraging computational predictions in order to improve literature-based GO annotation consistency and quality.
Other groups have previously investigated quality issues for GO annotations, using different strategies. For example, in his PhD dissertation, John MacMullen investigated variation between annotations from different curators and the factors that contributed to that variation, in a study of multiple model organism databases (27). Dolan et al. (28) developed a method for assessing GO curation consistency by comparing GO-slimmed literature-based annotations of orthologous gene pairs from mouse and human. Our method is novel in that it leverages two types of annotations: manually assigned annotations from the literature, and computational predictions. Annotations and predictions are sorted into pairs for a given gene and aspect, and the pairs may be categorized by the relationship between the GO terms used for each member of the pair. Here, we have presented the results of analysis of one such category: manual ‘unknown’ annotations paired with an InterPro signature-derived prediction. In the future, we will expand the analysis to include non-‘unknown’ literature-based annotations in SGD, and other sources of computational predictions in addition to InterPro, with the goal of developing a robust automated method for identifying and prioritizing literature-based GO annotations to review and update.
This type of analysis could also contribute to the quality of computational prediction methods, for example, by revealing areas of biology in which the prediction methods are less accurate and need to be adjusted. Discrepancies between literature-based annotations and computational predictions could also identify errors in mappings of InterPro signatures or E.C. numbers to GO, or errors and inconsistencies within the GO structure. Furthermore, the method should be applicable to GO annotations for any organism for which both literature-based and computational annotation is performed.
Since this method requires that both literature-based and computational annotations exist, we have here also addressed its limitations, by looking at the extent to which computational methods, specifically those developed by the GOA project, cover the entire genome. We found that computational predictions made by the GOA project cannot provide comprehensive annotation coverage for any of several disparate genomes that we surveyed. Even for the M. tuberculosis genome, where literature-based GO annotation is a small-scale effort, 13% of the genes that are currently represented in the GAF would lack annotation completely if no manual annotation were done. These results underscore the critical and ongoing need for literature-based curation to fill in these gaps, as well as to serve as the basis for comparison and improvement of prediction methods. For maximal quality of the biological information captured in genome databases, both manual and automated curation must co-exist. The method and results presented here demonstrate that rather than being alternative approaches to curation, manual annotation and computational prediction are complementary, and comparison of the two can result in the synergistic improvement of both.
Acknowledgements
We thank Karen Christie, Jodi Hirschman, and Marek Skrzypek for critically reading the manuscript; Karen Christie for work on a pilot project for this study; Catherine Ball, Harold Drabkin, Doug Howe, T.B.K. Reddy, Ralf Stephan, and Kimberly van Auken for assistance in determining counts of genes and annotations in other databases; and all members of the SGD project for helpful discussions and support.
Funding
National Human Genome Research Institute at the National Institutes of Health (HG001315 to J.M.C. and HG002273 to J.M.C., co-PI). Funding for open access charge: National Human Genome Research Institute at the National Institutes of Health (HG001315).
Conflict of interest. None declared.

{kind=link}
{kind=link}