





Abstract
In recent years, genomes from an increasing number of organisms have been sequenced, but their annotation remains a time-consuming process. The BioCyc databases offer a framework for the integrated analysis of metabolic networks. The Pathway tool software suite allows the automated construction of a database starting from an annotated genome, but it requires prior integration of all annotations into a specific summary file or into a GenBank file. To allow the easy creation and update of a BioCyc database starting from the multiple genome annotation resources available over time, we have developed an ad hoc data management system that we called Cyc Annotation Database System (CycADS). CycADS is centred on a specific database model and on a set of Java programs to import, filter and export relevant information. Data from GenBank and other annotation sources (including for example: KAAS, PRIAM, Blast2GO and PhylomeDB) are collected into a database to be subsequently filtered and extracted to generate a complete annotation file. This file is then used to build an enriched BioCyc database using the PathoLogic program of Pathway Tools. The CycADS pipeline for annotation management was used to build the AcypiCyc database for the pea aphid (Acyrthosiphon pisum) whose genome was recently sequenced. The AcypiCyc database webpage includes also, for comparative analyses, two other metabolic reconstruction BioCyc databases generated using CycADS: TricaCyc for Tribolium castaneum and DromeCyc for Drosophila melanogaster. Linked to its flexible design, CycADS offers a powerful software tool for the generation and regular updating of enriched BioCyc databases. The CycADS system is particularly suited for metabolic gene annotation and network reconstruction in newly sequenced genomes. Because of the uniform annotation used for metabolic network reconstruction, CycADS is particularly useful for comparative analysis of the metabolism of different organisms.
Database URL:http://www.cycadsys.org
Background
Next-generation sequencing technology and its many applications are revolutionizing genomics-based research (1, 2). Thanks to these novel approaches, the cost of obtaining the genome sequence of an organism is much reduced, and genome sequencing projects are being developed for many organisms. The good annotation of a genome is key for understanding the underlying biology (3). The assignment of specific functions to genes is a dynamic process; after the first automated computational analysis of all sequencing data (e.g. large expressed sequence tags (ESTs) collections, genomic DNA, single-gene characterization), further studies using both bioinformatics and experimental approaches are performed. General annotation information is collected in the GenBank database, but important data on gene function are also found in specialized dedicated databases. Among them, the BioCyc collection of Pathway/Genome Databases (PGDBs) constitutes an important resource for metabolism analyses.
The BioCyc databases are a type of Model Organism databases built using the Pathway Tools software system (4, 5). These databases include metabolic pathway data linked to genome information. Following the creation of EcoCyc for Escherichia coli (6, 7) and MetaCyc (a multi-organism database) (8, 9), the collection was later extended to another 160 organisms (10). This collection is in continuous expansion and, at the time of writing (October 2010), the collection contains 1004 PGDBs, classified in three categories according to the level of manual curation: 4 are intensively curated, 32 are computationally derived but subject to moderate curation and 968 are only computationally derived (http://biocyc.org/).
The quality of computationally derived BioCyc databases generated using the PathoLogic program of the Pathway Tools system is directly linked to the content of the annotation file used to build the database. This annotation file can be a GenBank flat file downloaded from an existing database (e.g. GenBank, FlyBase, etc.). For newly sequenced genomes, however, the gene or protein functional annotation is generally obtained using different methods and the annotation data are available in different formats.
It is important to have the latest annotation available in any given database. A key feature of the Pathway Tools system is to allow updates of the generated BioCyc database. The Pathway Tools makes this update in two ways: manually for each annotation update or importing an annotation file. Depending on the quantity of annotations to update and the source (or sources) of the update, keeping the latest annotation available in the BioCyc database can be demanding. The manual option is in fact not feasible in most cases, where a lot of new annotations need to be updated, and the management of annotation files can be difficult due to the lack of a specific annotation file generator.
Pathway Tools also offers a framework to perform a comparative analysis of the metabolism of different organisms. Nonetheless, when such analyses are performed using computationally derived metabolism reconstructions, the network comparison can be highly biased by the variable quality of the annotations available for different organisms.
To complement the Pathway Tools software and thus improve the generation of BioCyc databases, we developed CycADS: a data management system dedicated to the creation and update of Cyc databases. Our pipeline includes a SQL database and a set of Java programs for data exchange. CycADS allows the collection of annotations from different sources, the management of information over time and the easy output of collected data to computationally generate a BioCyc database with a higher information content. We tested our pipeline to develop two new databases: ‘AcypiCyc’ for the newly sequenced genome of the pea aphid (Acyrthosiphon pisum) (11) and ‘TricaCyc’ for the recently sequenced genome of the red flour beetle (Tribolium castaneum) (12). Furthermore, we also generated a new computationally derived BioCyc database for the Drosophila melanogaster (‘DromeCyc’).
In conclusion, CycADS helped the generation of improved BioCyc computationally derived databases, and allowed a global analysis of the metabolism of the pea aphid (11).
Implementation
The CycADS workflow
The workflow to use CycADS to generate a BioCyc database includes the following steps (Figure 1): CycADS can also be used to generate annotation files in other formats for use in different research applications (e.g. annotation management for microarray data analysis).
the genomic information (genes, RNA and proteins) is collected from GenBank (http://www.ncbi.nlm.nih.gov/Sitemap/samplerecord.html) and/or GFF3 (http://www.sequenceontology.org/gff3.shtml) flat files and stored in the database;
the protein functions are annotated using different methods that assign Enzyme Commission (EC) numbers and/or Gene Ontology (GO) identifiers;
all annotation information is gathered in the CycADS database, including complementary information about the method used for the functional assignment;
the annotations are then extracted from CycADS to generate flat files in a specific format (PF for PathoLogic File);
the PF files are loaded by the PathoLogic module of the Pathway Tools system to generate a BioCyc database for a given organism.
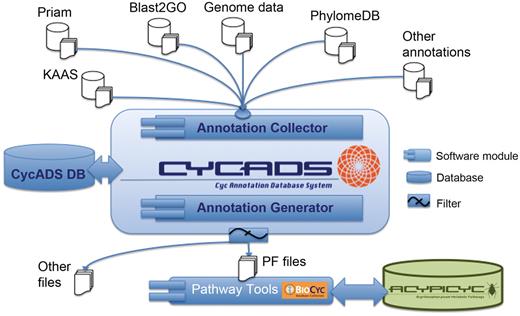
CycADS annotation management system workflow. Genomic information is combined in CycADS with the annotation data obtained using different methods and the collected data are filtered to produce the PathoLogic files (PF files output) that will then be used to generate the BioCyc databases with the Pathway Tools system (PathoLogic module). The annotations can also be extracted for other applications using the filtering system (other files output).
System overview
CycADS uses a specific SQL database model and a set of Java programs to import/export data. The system was tested in a MySQL database management system (DBMS) but it can be easily configured to use others SQL DBMS, like Oracle and PostgreSQL (see the manual at http://code.cycadsys.org).
The system Java code can be split in three layers:
‘Data’. Classes and interfaces to access and represent the entities used in the system. We can split this layer in two packages:
‘Database storage’. The classes responsible to store and retrieve the data to/from the repository. Currently, we are storing the data in a SQL database system and we are using SQL queries and SQL commands on the classes of this layer.
‘Database access’. The classes in this package represent logically the data and are the interface to access this layer. This package allows changing the database storage package (for example to use an Object Oriented DBMS) without modifying any class of the other layers. We developed this package with Java interfaces.
‘Logical’. This layer coordinates the commands requested, makes logical decisions and interacts with the data layer. It includes the information and logical rules to parse several different data format (to import or export) and to filter the exported data.
‘User interface’. This layer makes the interaction with the users. It gets the data provided by the users, triggers the logical processes and presents the data generated by the system. Currently, the user gives information to CycADS through the command line parameters and through the config.properties file, and the data are obtained from flat files.
The CycADS database
The CycADS database was designed to store the biological function data obtained for each protein from different annotation sources. In this database, we store all the data necessary to generate the metabolic network reconstruction of a given organism using Pathway Tools.
The logical database model, as shown in Supplementary Figure S1, contains the following entities:
‘Organism’: the organism analysed.
‘Sequence’: the chromosome or the contig for a given organism genome assembly (including the mandatory assembly version field). The objects of this entity may or may not include the ‘ACGT’ DNA sequence symbols.
‘Subsequence’: corresponds to one sequence fragment and it may or may not be continuous (i.e. if it is a gene it may or may not include introns).
‘DbxRef’: corresponds to an object in an external database. This entity has the following attributes: DBName and accession. DBName is a database external name or its abbreviation and accession is the identifier of this object in the external database. e.g. DBName = entrez-gene; accession = entrez-gene ID (for example: 100164132).
‘Annotation’: is the main entity in the database and is used when an annotation method m proposes to assign a qualifying note n to an object o. The annotation entity represents the proposition of assignment of n to o by m. The qualifying note n can be a simple text or a database entity object, like an external reference (DBxRef). According to the object types of n and o, we classify the annotation entities into the following annotation types:
‘SubseqAnnotation’: o is a subsequence.
Depending on the entity type of n, we have the following types of SubseqAnnotation objects: All annotation objects have the following attributes:
SubseqFeatureAnnotation: n is a feature (e.g. ‘gene’, ‘mRNA’, ‘CDS’, etc.).
SubseqDBxRefAnnotation: n is a DBxRef entity object (e.g. EC:4.1.1.15).
SubseqFunctionAnnotation: n is a functional text (e.g. ‘glutamate decarboxylase’).
‘DbxRefDBxRefAnnotation’: n and o are DBxRef entity objects. For example, this annotation type is used to relate an EC number to a KEGG orthology (KO) identifier.
‘Method’: indicates the method used to propose the assignment.
‘Score’: corresponds to the score assigned by the method used in the annotation process. In general, the score represents the reliability of the annotation as assessed by each single method. The attribute score is optional.
‘Parent’: indicates zero, one or more parent annotations. The parent annotations are the annotations supposed to be directly responsible for the existence of this annotation, for example, if the mRNA r comes from the Gene g then g is parent of r. The parent is used, for example, to get the gene name of a protein to generate a PF file.
An example of annotation is the following: if an annotation method m associates a CDS c to an EC number e with a score s, we create a SubseqDBxRefAnnotation object with the following attributes: method = m, object o = subsequence of c, qualifying note n = e, score = s and parent = null.
All database objects can have multiple synonyms stored as DBxRef objects. A synonym is the identifier of an object in an external database. Furthermore, all database objects may have several simple notes associated to them, that are generic values not representing an object in the database model (e.g. comments regarding an annotation).
Several public database schemas designed to store generic biological data exist: BioSQL (http://www.biosql.org), Chado (13,14), BioWareHouse (15), Atlas (16) and BioDWH (17). The BioWarehouse, Atlas and BioDWH are database schemas and toolkits to construct biological data warehouses. They were developed to retrieve and combine the data from many biological database sources, but they were not designed to store data from functional annotation processes. Moreover, the Atlas system is not currently available and the BioWarehouse was designed for prokaryotic organisms data and has some limitations when applied to data from eukaryotes. The Chado and BioSQL schemas are generic and were designed to be used with many different kinds of data and applications. We tested the BioSQL schema, but it did not precisely fit to our needs, mainly because our annotation entities refer to three other entities (qualifying note n, object o and the annotation method m). If we had used one of these cited schemas, we would have had to make many changes to the schema to adapt to our needs or we would have had to store our data in a schema with weak consistency rules in the RDBMS.
We therefore created a specific SQL database schema to implement our logical database model providing more relational integrity to the system.
Annotation collector module
The CycADS system includes loading programs developed to collect data from different file formats. The Annotation Collector module can be easily configured, using specific parameters in the configuration file, to fit many different data file formats. These parameters are very important to allow a quick adaptation to changes in the files to load. Some flat file formats, like GenBank and GFF3, are very flexible and allow the generator of the file to store some information in generic and non-standardized fields (or tags) of the file. CycADS can be configured to import the information from these not standardized fields.
At present, CycADS contains programs to load files with a GenBank, GFF3 or text column format. In general, a text file with columns is used to store either the relationship between objects or the link between different identifiers of the same object. CycADS can import the data in these files either as annotations or as synonyms. The annotations are used to represent relationships between different objects, like to assign a GO function to a protein sequence. The synonyms are used to represent links between different identifiers of the same object, such as to link a GI number to an mRNA sequence (for a detailed description of each collector program, see the manual at: http://code.cycadsys.org).
Annotation generator module
CycADS was developed to generate a specific output file format: the PF file used by the PathoLogic program in Pathway Tools to create a BioCyc database (4, 5). Nonetheless, the annotation generator parameters can be easily adjusted to output the data in other formats.
As for the loading programs, the generator program is flexible and can be configured to filter and generate the output file containing information from different entities as needed. For example, the DBLink field in the PF file can be filtered to contain DBxRefs only from specific external databases (e.g. GenBank, PhylomeDB, etc.).
Each assignment of an EC (or GO) number to a protein has a score generated by CycADS during the extraction step. We therefore say that each EC (or GO) annotation has a CycADS extraction score. One protein can have zero, one or many EC (or GO) annotations. At present, the CycADS extraction score is the quantity of methods that assign the EC (or GO) number to the protein, or in other words, the number of methods that agree with a given EC (or GO) annotation. Nonetheless, with a simple change in the configuration file, CycADS can also assign specific weights to each annotation method and use this in the final filtering system. Thus, for example, CycADS can generate a PF file that would exclude a specific method for which we have assigned a weight zero.
One important feature in this module is the use of a filter based on the CycADS extraction score to generate the EC and GO annotations of the proteins. Thus, CycADS can produce a PF file (and consequently a BioCyc database) with only the EC (or GO) annotations with a CycADS extraction score above a threshold chosen by the user.
CycADS user interface
The CycADS programs are executed by command line and get supplementary parameters from a configuration file (for details, see the manual at: http://code.cycadsys.org).
Results
From CycADS to AcypiCyc, TricaCyc and DromeCyc
CycADS generated successfully the BioCyc databases for the pea aphid A. pisum (AcypiCyc), the red flour beetle T. castaneum (TricaCyc) and the fruit fly D. melanogaster (DromeCyc).
The generation of AcypiCyc and TricaCyc using CycADS was performed to test the reliability of our pipeline for the generation of BioCyc databases for different organisms whose genomes have been recently sequenced. On the other hand, DromeCyc was produced to test CycADS in the generation of a BioCyc database for a well annotated genome.
Our goals (successfully achieved) in the generation of these BioCyc databases were:
to enrich the PathoLogic file (and consequently the BioCyc database) with the maximum information available;
to filter out undesired information from the BioCyc database (e.g. inconsistent or untrusted EC and GO annotations); and
to easily update the generated BioCyc database every time new annotation data become available.
Data
Genome and protein sequences
The genome information (CDS, RNA and gene descriptions) to generate AcypiCyc, TricaCyc and DromeCyc was obtained from GenBank and GFF files. These files were downloaded from the NCBI site and/or from the organism-dedicated databases: AphidBase (18) (http://www.aphidbase.com/aphidbase/), BeetleBase (19) (http://beetlebase.org/) and Flybase (20) (http://flybase.org/). For A. pisum and T. castaneum, we used the GFF files coming from the corresponding Chado genome databases. For D. melanogaster, we used two GenBank files (one from the NCBI and one from FlyBase) to obtain genome information and annotations.
For the organisms A. pisum (AcypiCyc) and T. castaneum (TricaCyc), the protein sequence for the annotation process (described below) was downloaded as amino acid sequence FASTA files from the organism-specific databases.
EC annotations
The EC number annotations for all genes were obtained for AcypiCyc and TricaCyc using three methods: the KEGG Automated Annotation System (KAAS) (21), Blast2GO (22) and PRIAM (23).
The KAAS annotation was performed using the online ‘KAAS-KEGG Automatic Annotation Server’ (http://www.genome.jp/tools/kaas/) with the full genome option BBH (bi-directional best hit) method to assign orthologs (KO identifiers). Two executions of the KAAS method were done with different pre-selected datasets of species: ‘for Eukaryotes’ and ‘for GENES’. The output files with the mappings protein-KO are in tabular column text format. The EC numbers were obtained using the information present in the KO definitions file (http://www.genome.jp/kegg/).
The Blast2GO (http://www.blast2go.org/) analysis (Blast2GO-EC method) included three steps: (i) protein sequence Blast analysis with default settings, (ii) GO identifiers assignment using the GO mapping module, and (iii) EC numbers assignment by the enzyme mapping module (both steps (ii) and (iii) were performed using the Blast2GO default parameters). The output files with the mappings protein-EC are in tabular column text format.
Using PRIAM (http://priam.prabi.fr/), each sequence was compared to all PRIAM profiles (domains) and for each protein, the output was the EC number(s) of all the profiles that matched with a maximal e-value of 10−3 and a minimal proportion of 70% of the domain length matching (default values for the PRIAM input parameters). The output files with the mappings protein-EC are in tabular column text format.
The EC numbers for the construction of DromeCyc were obtained from the GenBank file annotations. This choice is motivated by the fact that most of the information gathered using the different annotation methods relies heavily on the information of the D. melanogaster genome to assign a function.
GO annotations
The GO annotations presented in AcypiCyc and TricaCyc were assigned to genes using the PhylomeDB method. This assignment took advantage of the phylogenomic analysis performed for A. pisum and T. Castaneum using the PhylomeDB pipeline (24,25). The GO annotations from the D. melanogaster genes were transferred to the orthologous pea aphid genes and a score (or evidence level) was assigned to each annotation depending on two factors: type of orthology relationship and conservation of annotation in ancestral nodes. In brief, the type of orthology relationship takes into account whether the source and target sequences are one-to-one orthologs; in this scenario the transfer of a functional annotation is most safe, since there is an absence of recent duplications that may have involved processes of functional change. Transfers among one-to-one orthologs are thus given a higher score (+1). In contrast, other types of orthology relationships (one-to-many and many-to-many) that are more likely to underlie processes of functional change are given a lower confidence score (+0). The second factor that is taken into account is the presence of the same annotation in an ortholog from any common outgroup of the considered source and target sequences. If this is the case, the function is assumed to be conserved across large evolutionary distance, including the common ancestors of the considered sequences. Thus annotations that fulfil this condition are given a higher score (+1), than others (+0). A third factor that is not relevant for this case, regards the phylogenetic distance between the source and the target species. In both A. pisum and T. castaneum, the distance to the source species (D. melanogaster) receives the same score (+1). Thus scores assigned to GO annotations range from 1 (there is a one-to-many, or many-to-many ortholog in D. melanogaster with that annotation) to 3 (there is a one-to-one ortholog in D. melanogaster with that function, which is also conserved in orthologs from out-group species) in the two insect genomes considered here. All orthology predictions and functional transfers were computed using the species overlap algorithm as implemented in ETE 2.0 (26). In brief, this is an automated, phylogeny-based algorithm (27) that analyses each gene-tree topology while assigning a duplication event at nodes where the two daughter partitions share at least a species, and speciation events when no evidence for species overlap is found. Orthology and paralogy predictions are then predicted according to the original definition of orthology (i.e. orthology if the ancestral node is an inferred speciation event, and paralogy if the ancestral node is inferred as a duplication event) (28).
The output files with the mappings protein-GO are in tabular column text format and for T. castaneum a newer format file was used with more information than the file used for A. pisum. This allowed us to test and show the flexibility of the CycADS system when working with different format files, even coming from a same source. The GOs present in DromeCyc were directly extracted from the GO annotation file downloaded from FlyBase.
A summary of the annotation used for the construction of each individual database is presented in Table 1.
Annotation methods by database
| EC annotation | No. | GO annotations | No. | |
|---|---|---|---|---|
| AcypiCyc | KAAS(2), PRIAM, Blast2GO | 4 | Phlylome DB inference | 3 |
| TricaCyc | KAAS(2), PRIAM, Blast2GO | 4 | Phlylome DB inference | 3 |
| DromeCyc | GenBank from NCBI | 1 | GO from FlyBase | 1 |
A summary table including the annotation methods used for each database (at the time of publication). For the EC numbers: ‘KAAS’—two different KAAS methods were used to annotate the protein sequence, using two different reference datasets (Eukaryotes and GENES, see text for details); ‘PRIAM’—the annotation was performed using default parameters; ‘Blast2GO’—inference of EC number from the GO annotation; ‘GenBank file from NCBI’—downloded file. For the GO numbers: the ‘Phylome DB inference’ with three levels of confidence (see text for details); ‘GO from FlyBase’—downloaded file.
The generation of BioCyc databases
Using the Annotation Collector module, we successfully loaded in the CycADS database the genome and annotation data described before.
We produced six online (available at http://acypicyc.cycadsys.org) BioCyc databases: two for the pea aphid A. pisum (‘AcypiCyc All by CycADS’ and ‘AcypiCyc Filtered by CycADS’), two for the red flour beetle T. castaneum (‘TricaCyc All by CycADS’ and ‘TricaCyc Filtered by CycADS’) and two for the fruit fly D. melanogaster (‘DromeCyc by CycADS’ and ‘DmeBioCyc by the Genbank-PathwayTools’).
To show the potential use of the CycADS extraction score (described in the ‘Implementation’ section), we generated different versions for A. pisum and T. castaneum, with two different annotation reliability levels for each of them (‘all’ and ‘filtered’ versions). To generate these different versions, we used two separate PF files for each organism: (i) a fully unfiltered version (for the ‘all’ version) where one annotation method for either EC or GO is sufficient to assign the respective annotation to the protein; (ii) a more strictly filtered version (for the ‘filtered’ version) where an EC annotation is assigned to a protein only if all four EC annotation methods agree, and a GO annotation is assigned to a protein only if the GO annotation has three evidence levels in the PhylomeDB-based method. All possible combinations of annotation scores for EC and GO can be used to generate multiple BioCyc databases very easily, thanks to CycADS. Many of these possible combinations were used to perform the comparative annotation analysis in AcypiCyc, as described in details below (see Supplementary Table S1).
We also generated two databases for the model organism D. melanogaster using a specific procedure. Two BioCyc database versions were generated: (i) DromeCyc by CycADS: using the CycADS pipeline to combine two different and complementary GenBank files (from the NCBI and FlyBase databases) and the GO annotations coming from FlyBase; (ii) DmeBioCyc by the GenBank-PathwayTools: straight from the NCBI GenBank data file using the classical GenBank pipeline of the Pathway Tools software. While writing this article, a new Tier 2 database was developed by another group for D. melanogaster (FlyCyc), that is available for comparison with AcypiCyc. Even if FlyCyc is manually curated and DromeCyc (by CycADS) only computationally derived the two databases are not very different from the annotation point of view. In fact, the large majority (89%–738/830) of EC numbers identified are common (21 and 71 are unique, respectively, to DromeCyc and FlyCyc) betweeen the two databases.
Even if AcypiCyc, TricaCyc and DromeCyc were generated using the PF file Pathway Tools pipeline, several features coming from CycADS distinguish them from that of a ‘classical’ BioCyc computationally derived database (BioCyc Tier 3). In fact, any database generated using CycADS includes, for each annotated protein, more detailed information about the source of the annotation in the Summary of the Gene webpage (see Figure 2). In both versions (‘all’ and ‘filtered’ versions) of AcypiCyc and TricaCyc, the information about the annotation method and the CycADS extraction score for each EC and GO annotation can be found in the gene page as shown in Figure 2, while in the metabolism reconstruction only the EC and GO annotations above the chosen cutoff are taken into account.
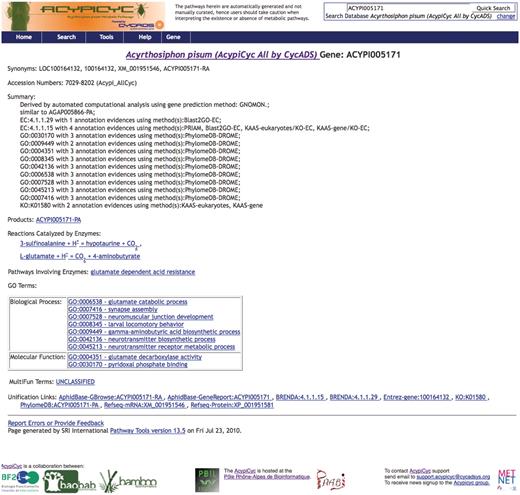
Screenshots of a BioCyc database generated by CycADS. An example page from AcypiCyc showing the enrichment of a BioCyc gene page with complementary information about the annotation source included in the ‘Summary’ and extra hyperlinks (‘Unification Links’) to important resources.
DromeCyc generated by CycADS data integration provides more information than the DmeBioCyc version. In particular, DromeCyc offers complementary information (e.g. protein and gene synonyms) that are not loaded in the DmeBioCyc by the GenBank-PathwayTools.
This enriched version is achieved thanks to the automated integration of multiple sources that allowed us, for example, to include in the DromeCyc gene pages the GO evidence source code (http://www.geneontology.org/GO.evidence.shtml) obtained from the FlyBase GO annotation file. The details in EC/GO annotations enables researchers to evaluate the source method.
Using CycADS, all our databases have also more external links in the gene-specific page than a version generated with the straight upload pipeline of Pathway Tools. These additional hyperlinks include, for example, links to organism-unique resources (AphidBase, BeetleBase and FlyBase in the presented work) and to other sources of information about the genes, such as phylogeny (e.g. PhylomeDB).
This useful extra-links feature is particularly evident when comparing the two versions for D. melanogaster. In the case of A. pisum, thanks to the collaborations as part of the International Aphid Genomics Consortium, a link back to AcypiCyc is present in the outer database (AphidBase) allowing a researcher to move easily among the different resources.
Contribution of the different annotation methods: the AcypiCyc example
To evaluate the value of compiling different annotation methods, we used CycADS to generate several versions of the AcypiCyc database, using different cut-offs to the EC and GO annotation evidences (see Supplementary Table S1). Only two of these databases are available online (‘AcypiCyc All by CycADS’ and ‘AcypiCyc Filtered by Cycads’). We compared the number of reactions and annotated genes (catalysing at least one reaction) identified by the different annotation methods.
From comparison, we verified that each method contributes to the annotation of many distinct genes and reactions. For example, 902 reactions were identified as present by either all four EC annotation methods and/or the highest level of confidence of the PhylomeDB method for the GO annotation (‘AcypiCyc Filtered by Cycads’), while 1622 by at least one single annotation method for EC or GO (‘AcypiCyc All by CycADS’). Thus depending on the level of reliability wanted, the user can eliminate up to 44.4% of the reactions present in the ‘all’ database version.
To compare the results of the different annotation methods in greater details, we concentrated on the annotation assigning an EC number to the proteins, as we have only the PhylomeDB method for the GO assignment (even if different levels of confidence are present in the method). In Figure 3, a Venn-diagram summary of the comparisons shows the relative contributions of the different annotation methods used. This type of comparison could also be used to evaluate the relative contributions of each method to obtain the desired cut-off level for a metabolic network reconstruction. In fact, the different contributions of each method show that a real network comparison needs to take into account the annotation source for a given genome. Indeed, only 428 reactions (435 genes) are annotated by all EC methods, giving a high level of support for these reactions in the network. For many other cases, different methods do not totally agree, so even if there is a partial overlap, the summary contributions can be of interest. The relatively weak overlap can be partially explained by the different approaches used by the annotation method. The slightly higher overlap between KAAS and PRIAM can be linked to the fact that both methods, even if using different annotation approaches, are geared towards enzyme specific annotation based on sequence similarity, while in Blast2GO the EC numbers are inferred from a global annotation of GO terms.
![Comparison of the EC annotation by different methods in AcypiCyc. (A) Reaction annotation by EC methods. Venn-diagrams showing the number of reactions (total of 1176) identified in the metabolic reconstructions using data from the different annotation methods [PRIAM, KAAS (two methods), Blast2GO-EC], the total number of reactions annotated by each method is specified in black below the method name, while specified in white is the number of unique or shared reactions among annotations. (B) Gene annotation by EC methods. Venn-diagrams showing the number of genes (total of 2281) annotated using the different methods [colour code for annotations as in (A)]. Note: multiple genes may catalyse a single reaction. This figure was generated using Aduna Cluster Map - http://www.aduna-software.com/technology/clustermap.](https://oupdevcdn.silverchair-staging.com/oup/backfile/Content_public/Journal/database/2011/10.1093_database_bar008/1/m_bar008f3.jpeg?Expires=1787886184&Signature=JhsSpR30PZYHWi-8EU8Ececmz46-3C1C0ybD-VGCWbjyAagxzhmizMBKUqmn4ns3Axa9R2PYNBC9R46effZnavpeiREp~l7pVtfoYk7xqjGfLyF5faho2I~ltKga37VNAdVZoELLqin3ysJDfrtBinN6BIA1fsbYucSQIjKvBuFsdtIjWp9j-5Y79DHNgsCgfh5UAxBnmvR6DxMX5H-6eptxRz-8GUX9ZK1ZHNpbtBLlle--~HB6T5lP7CmmVfWpk2~1lNfTLCfhYCuccNg32-tYSZ1sm5DjxRCqKudFTMoYeSPL4xJiphgjynSwX537NLIFgkLJ-eL289K9ZytA4w__&Key-Pair-Id=APKAIYYTVHKX7JZB5EAA)
Comparison of the EC annotation by different methods in AcypiCyc. (A) Reaction annotation by EC methods. Venn-diagrams showing the number of reactions (total of 1176) identified in the metabolic reconstructions using data from the different annotation methods [PRIAM, KAAS (two methods), Blast2GO-EC], the total number of reactions annotated by each method is specified in black below the method name, while specified in white is the number of unique or shared reactions among annotations. (B) Gene annotation by EC methods. Venn-diagrams showing the number of genes (total of 2281) annotated using the different methods [colour code for annotations as in (A)]. Note: multiple genes may catalyse a single reaction. This figure was generated using Aduna Cluster Map - http://www.aduna-software.com/technology/clustermap.
From the analysis of the contributions of the different annotations, it is also important to observe that GO assignment using the PhylomeDB method added several complementary reactions. Indeed, 60 new reactions were added using the low cut-off of 1 evidence (by PhylomeDB), showing the usefulness of such complementary phylogenenetic annotation method.
Discussion
Annotation is key to understanding the biology underlying the genome of an organism (3). The ever-ongoing genome annotation process depends critically on newly developed ad hoc tools for different applications. Metabolic network reconstructions and analyses are only as good as the annotation. In response to this changing research scenario, the CycADS annotation database system allows easy and automated annotation integration over time using the BioCyc powerful metabolism reconstruction and visualization tools, thus complementing the Pathway Tools software and BioCyc Pathways/Genome Databases (PGDB). Such constant update and quality checking of annotations is crucial for successful downstream analyses of metabolism network and will permit continuous access to up to date information.
The quality of the annotation is bound to improve over time and the sequences of multiple genomes will allow for an enrichment of the information available for several organisms thanks to the work of multiple scientific communities. The availability of more and more genome sequences will also open the way to a comparative analysis of metabolism. To perform such studies, an ‘homogeneous’ gene annotation is very important. The CycADS annotation evidence filter based on a score is a first step towards better consistency between different organisms. The CycADS annotation filtering system allows the users to test different confidence levels of annotation and to perform network comparisons between organisms using the same annotation evidence threshold. Even if the setting of a cut-off annotation level does not guarantee the reality of the reactions present in the metabolic network, comparing different organisms using the same kind and level of annotation can alleviate the problems posed by variable annotation quality on the results of the comparative analyses. Thus, even if it is impossible to assess the right evidence level purely based on an in silico annotation, CycADS enables the user to compare networks based on a similar level of functional annotation quality.
A substantial amount of work for the primary development of CycADS was needed due to the differences in the format of the data to import. CycADS can be easily modified to accommodate different data sources and used for metabolism annotation of other organisms with newly sequenced genomes. The AcypiCyc database developed using CycADS has allowed us to perform a global evaluation of the pea aphid metabolic capabilities (11), with particular attention to the amino acid metabolism due to the importance of these pathways in the symbiosis between the pea aphid and the bacteria Buchnera aphidicola (29). The comparison of the results obtained using CycADS and manual annotation for these metabolic pathways has demonstrated a good performance of the automated annotation used in AcypiCyc [see Supplementary Table in ref. (29)]. Thus AcypiCyc is a key resource for computational systems biology research to analyze the integrated metabolic network shared between the aphid and its symbiotic bacterium, for which a BioCyc database already exists. The development of BioCyc databases for D. melanogaster and T. castaneum allows the use of comparative analysis tools in AcypiCyc website to compare the metabolism of these insects to that of the pea aphid.
In particular, the annotations of A. pisum and T. castaneum were performed with the same annotation methods and CycADS extraction score; this allows an annotation-consistent comparison of the metabolic networks of these two insects. The extension to multiple other organisms, depending on the research questions, will be greatly facilitated by the CycADS system. The development of an even better integration with the Pathway Tools software allowing bidirectional exchange of information about the network annotations could help improve the downstream analyses.
Conclusions
CycADS is an integrated software and database system for the management of annotation information formatted for easier generation of enriched computationally derived BioCyc databases. The availability of an increasing number of fully sequenced genomes for multiple organisms will allow comparative analysis of metabolic network. In this arena, the harmonization of annotation information offered by CycADS offers a valuable key for data management.
Availability and requirements
Project name: CycADS
Project home page: http://www.cycadsys.org
Operating systems: Windows, Linux or Mac OS X
Programming languages: Java, SQL
Other requirements: Java JDK 6
License: GNU
Contact: support@cycadsys.org
Funding
Agence National de la Recherche (ANR, France); and the Biotechnology and Biological Sciences Research Council (BBSRC, UK) [MetNet4SysBio project (http://www.metnet4sysbio.org/) to A.E.D., M-F.S. and H.C.]; the Spanish Ministry of Science and Innovation [BFU2009-09168 and GEN2006-27784E to T.G.]; ‘NSF research grant [IOS-0919765 to A.E.D.]; and The Sarkaria Institute of Insect Physiology and Toxicology [to A.E.D]. Funding for open access charge: ANR-BBSRC MetNet4SysBio.
Conflict of interest. None declared.
Acknowledgements
The authors would like to thank the staff of the Pôle Rhône-Alpes de Bioinformatique, PRABI (www.prabi.fr) in Villeurbanne (France) for the support in the installation of the Pathway Tools software (BioCyc) and for the day-to-day maintenance of the servers hosting AcypiCyc. Thanks also to Thomas Bernard for his help with the new version of PRIAM. The authors would also like to thank the reviewers for their valuable comments that improved the article. A.F.V, M-F.S., H.C. and S.C. planned the database and software development. A.F.V and S.C. oversaw the project development. A.F.V. wrote the code and designed the database. L.C. and P.B.P. contributed to updates in the code and to the design updates. A.F.V., S.C., P.B.P. and A.S.V. performed the EC annotations. A.S.V. performed the AcypiCyc annotation detailed analysis. J.H-C and T.G. extracted the GO annotation from PhylomeDB and developed the GO assignment scoring system. G.F., F.C., Y.R. and A.E.D. provided expert online testing for the AcypiCyc database. S.C. and A.F.V. organized the manuscript writing integrating contributions from A.S.V., J.H.C., T.G. and P.B.P. All authors revised and approved the manuscript.
The original version of this manuscript was incorrect. The correspondence email for Augusto F. Vellozo was erroneous and has now been corrected.
References
Author notes
Present address: Amélie S. Véron, INSERM UMR590, Centre de Recherche en Cancérologie de Lyon (CRCL), Centre Léon Bérard, 28 rue Laënnec, F-69008, Lyon, France.
Present address: Ludovic Cottret, INRA, UMR792 Ingénierie des Systèmes Biologiques et des Procédés, Toulouse, France.

{kind=link}
{kind=link}
{kind=link}