





Abstract
Gene function curation via Gene Ontology (GO) annotation is a common task among Model Organism Database groups. Owing to its manual nature, this task is considered one of the bottlenecks in literature curation. There have been many previous attempts at automatic identification of GO terms and supporting information from full text. However, few systems have delivered an accuracy that is comparable with humans. One recognized challenge in developing such systems is the lack of marked sentence-level evidence text that provides the basis for making GO annotations. We aim to create a corpus that includes the GO evidence text along with the three core elements of GO annotations: (i) a gene or gene product, (ii) a GO term and (iii) a GO evidence code. To ensure our results are consistent with real-life GO data, we recruited eight professional GO curators and asked them to follow their routine GO annotation protocols. Our annotators marked up more than 5000 text passages in 200 articles for 1356 distinct GO terms. For evidence sentence selection, the inter-annotator agreement (IAA) results are 9.3% (strict) and 42.7% (relaxed) in F 1 -measures. For GO term selection, the IAAs are 47% (strict) and 62.9% (hierarchical). Our corpus analysis further shows that abstracts contain ∼10% of relevant evidence sentences and 30% distinct GO terms, while the Results/Experiment section has nearly 60% relevant sentences and >70% GO terms. Further, of those evidence sentences found in abstracts, less than one-third contain enough experimental detail to fulfill the three core criteria of a GO annotation. This result demonstrates the need of using full-text articles for text mining GO annotations. Through its use at the BioCreative IV GO (BC4GO) task, we expect our corpus to become a valuable resource for the BioNLP research community.
Database URL:http://www.biocreative.org/resources/corpora/bc-iv-go-task-corpus/ .
Introduction
The Gene Ontology (GO; http://www.geneontology.org ) is a controlled vocabulary for standardizing the description of gene and gene product attributes across species and databases ( 1 ). Currently, there are about 40 000 GO terms that are organized in a hierarchical manner under three GO sub-ontologies: Molecular Function, Biological Process and Cellular Component. Since its inception, GO terms have been used in more than 126 million annotations to more than 9 million gene products as of January 2013 ( 2 ). The accumulated GO annotations have been shown to be increasingly important in an array of different areas of biological research ranging from high-throughput omics data analysis to the detailed study of mechanisms of developmental biology ( 3–6 ).
Among the 126 million GO annotations, most are derived from automated techniques such as mapping of GO terms to protein domains and motifs (InterPro2GO) ( 7 ) or corresponding concepts in one of the controlled vocabularies maintained by UniProt ( 8 ); only a small portion (<1%) are derived from manual curation of published experimental results in the biomedical literature ( 2 ). While the former approach is efficient in assigning large-scale higher-level GO terms, the latter provides experimentally supported, more granular GO annotations that are critical for the kinds of analyses mentioned above. Generally speaking, the manual GO annotation process first involves the retrieval of relevant publications. Once found, the full-text is manually inspected to identify the gene product of interest, the relevant GO terms and the evidence code to indicate the type of supporting evidence, e.g. mutant phenotype or genetic interaction, for inferring the relationship between a gene product and a GO term. Such a process is time-consuming and labor intensive, and thus, many model organism databases (MODs) are confronted with a daunting backlog of GO annotation. For instance, in recent years, the curation team of the Arabidopsis Information Resource (TAIR) has been able to curate only a fraction of newly published articles that contain information about Arabidopsis genes (<30%) ( 9 ). It is thus clear that the manual curation process requires computer assistance, and this is evidenced by a growing interest in, and need for, semiautomated or fully automated GO curation pipelines ( 9–19 ). In particular, a number of studies ( 20–28 ) have attempted to (semi) automatically predict GO terms from text including a previous BioCreative challenge task ( 29 ). However, few studies have proven useful for assisting real-world GO curation. Based on a recent study, enhanced text-mining capabilities to automatically recognize GO terms from full text remains one of the most in-demand tasks among the biocuration community ( 30 ).
As concluded in the previous BioCreative task ( 29 , 31 ), one of the main difficulties in developing reliable text-mining applications for GO curation was ‘the lack of a high-quality training set consisting in the annotation of relevant text passages’. Such a sentence-level annotation provides in practice the evidence human curators use to make associated GO annotations. To advance the development of automatic systems for GO curation, we propose to create a corpus that includes the GO evidence text along with three essential elements of GO annotations: (i) a gene or gene product (e.g. Gene ID: 3565051, lin-26), (ii) a GO term (e.g. GO:0006898, receptor-mediated endocytosis) and (iii) a GO evidence code [e.g. Inferred from Mutant Phenotype (IMP)]. There are some challenges associated to creating such a corpus: the evidence texts for GO annotations may be derived from a single sentence, or multiple continuous, or discontinuous, sentences. The evidence for a GO annotation could also be derived from multiple lines of experimentation, leading to multiple text passages in a paper supporting the same annotation. In addition, as many learning-based text-mining algorithms rely on both positive and negative training instances, it is therefore important to capture all of the curation-relevant sentences to ensure the positive and negative sets are as distinct as possible. The usefulness of such evidence sentences has been demonstrated in previous studies such as mining protein–protein interactions from the bibliome ( 32 , 33 ).
The exhaustive capture of evidence text in full-text articles makes our data set, namely, the BioCreative IV GO (BC4GO) corpus, unique among the many previously annotated corpora [e.g. ( 34–38 )] for the BioNLP research community. To our best knowledge, BC4GO is the only publicly available corpus that contains textual annotation of GO terms in accordance with the general practice of GO annotation ( 39 ) by professional GO curators. For instance, while in a previous study ( 17 ) every mention related to a GO concept was annotated, in BC4GO we have annotated only those GO terms that represent experimental findings in a given full-text paper.
Methods and materials
Annotators
Through the BioCreative IV User Advisory Group, we recruited eight experienced curators from five different MODs: FlyBase ( http://flybase.org/ ) (two curators), Maize Genetics and Genomics Database (MaizeGDB) ( http://www.maizegdb.org/ ) (one curator), Rat Genome Database (RGD) ( http://rgd.mcw.edu/ ) (three curators), TAIR ( http://www.arabidopsis.org/ ) (one curator) and WormBase ( http://www.wormbase.org/ ) (one curator). All our annotations were performed according to the Gene Ontology Consortium annotation guidelines ( http://www.geneontol ogy.org/GO.annotation.shtml ).
Annotation guidelines
For achieving consistent annotations between annotators, the task organizers followed the usual practice of corpus annotation ( 34–37 , 40 , 41 ), which is also a GO annotation standard: first we drafted a set of annotation guidelines and then asked each of our annotators to follow them on a shared article as part of the training process. The results of their annotations on the common article were shared among all annotators and subsequently the discrepancies in their annotations were discussed. Based on the discussion, the annotation guidelines were revised accordingly. For brevity, we only discuss below the two kinds of evidence text passages we chose to capture. The detailed guidelines are publicly available at the corpus download website.
Experiment type
These sentences describe experimental results and can be used to make a complete GO annotation (i.e. the entity being annotated, GO term and GO evidence code). The annotation of such sentences is required throughout the paper, including the abstract, and any supporting summary paragraphs such as ‘Author summary’ or ‘Conclusions’.
On the other hand, the amount of UNC-60B-GFP was reduced and UNC-60A-type mRNAs, UNC60A-RFP and UNC-60A-Experiment were detected in asd-2 and sup-12 mutants ( Figure 2 H, lanes 2 and 3), consistent with their color phenotypes shown in Figure 2 C and 2 A, respectively. (PMC3469465)
This sentence contains information about the following:
The gene/protein entities: asd-2 and sup-12
GO term: regulation of alternative mRNA splicing, via spliceosome (GO:0000381)
GO evidence code: IMP
Summary type
Distinct from statements that describe the details of experimental findings, papers also include many statements that summarize these findings. These summary statements do not necessarily indicate exactly ‘how’ the information was discovered, but often contain concise language about ‘what’ was discovered. Such sentences are helpful to capture because they may inform GO term selection in a concise manner despite the lack of information about evidence code selection.
Example 2: Taken together, our results demonstrate that muscle-specific splicing factors ASD-2 and SUP-12 cooperatively promote muscle-specific processing of the unc-60 gene, and provide insight into the mechanisms of complex pre-mRNA processing; combinatorial regulation of a single splice site by two tissue-specific splicing regulators determines the binary fate of the entire transcript. (PMC3469465)
The gene/protein entities: ASD-2 and SUP-12
GO term: regulation of alternative mRNA splicing, via spliceosome (GO:0000381)
GO evidence code: N/A
Article selection
The 200 articles in the BC4GO corpus are chosen from annotators’ normal curation pipelines at their respective MODs. Such a protocol minimizes the additional workload to our curators while at the same time guarantees the curated papers are representative of real-life GO annotations and reflect a variety of biological topics. Another requirement is that annotated articles are published in a list of select journals (e.g. PLoS Genetics) in PubMed Central (PMC) that allow free access and text analysis.
Annotation tool
A web-based annotation tool, developed by J.D., K.V.A., H.M.M. and P.W.S. for use in the annotation process, is shown in Figure 1 . The tool allows the upload of full-text articles in either HTML or XML formats and subsequently displays the article in a Web browser. Currently, the tool allows the annotator to select and highlight a single sentence, or multiple sentences (regardless of whether they are contiguous or not) as GO evidence text. When a sentence is highlighted, a pop-up window appears for annotators to enter required GO annotation information: a GO term, a GO evidence code and associated gene(s). The tool also allows the annotators to preview their annotations before committing them to the database. Annotation results of each paper can be downloaded as HTML files as well as in a spreadsheet (XLXS format).
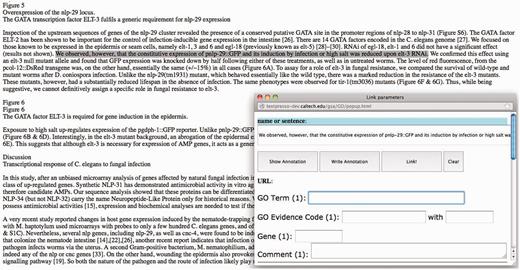
Screenshot of the GO annotation tool. When a line or more of text is highlighted, a pop-up window appears where annotation data are entered.
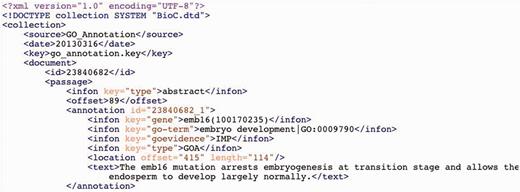
A sample of GO annotation in BioC format.
Post-challenge analysis: inter-annotator agreement
To gain insight on the consistency of annotation results and assess the difficulty of manually annotating text for GO annotation, two curators from RGD (S.J.F.L., G.T.H.) agreed to re-annotate a separate subset of 10 papers. Each of them did blind annotation of 10 papers from the training and development sets that have been annotated by another curator. This provided a set of 20 papers for calculating inter-annotator agreements (IAAs).
Final data dissemination
Both full-text articles and associated GO annotations (downloaded from PMC and the annotation tool, respectively) were further processed before releasing to the BC4GO task participants. Specifically, we chose to format our data using the recently developed BioC standard for improved interoperability ( 44 ). First, for the 200 full-text articles, we converted their XMLs from the PMC format to the BioC format. Next, we extracted annotated sentences from downloaded HTML files and identified their offsets in the generated BioC XML files. Finally, for each article we created a corresponding BioC XML file for the associated GO annotations. Figure 2 shows a snapshot of our final released annotation files where one complete GO annotation is presented using the BioC format. For the gene entity, we provide both the gene mention as it appeared in the text and its corresponding NCBI gene identifier.
Results and discussion
Corpus statistics
The task participants are provided with three data sets comprising 200 full-text articles. The training set of 100 curator-annotated papers was intended to be used by task participants for developing their algorithms or methods. Similarly, the development data set (50 papers) was to be used for additional training and validation of methods. The test set data (another 50 papers) was to be used strictly for evaluating the final performance of the different methods. Table 1 shows the number of articles curated by each MOD for each data set. On average, each curator contributed ∼25 articles for the task during this period.
Number of curated articles per MOD
| Data set | FlyBase | MaizeGDB | RGD | TAIR | WormBase | Total |
|---|---|---|---|---|---|---|
| Training set | 19 | 21 | 43 | 10 | 7 | 100 |
| Development set | 8 | 5 | 25 | 4 | 8 | 50 |
| Test set | 12 | 4 | 20 | 7 | 7 | 50 |
| Subtotal per team | 39 | 30 | 88 | 21 | 22 | 200 |
Table 2 shows the main characteristics of the BC4GO corpus. Each annotation includes four elements: the gene/protein entity, GO term, GO evidence code and evidence text (See Figure 2 ). Note that one text passage can often provide evidence for annotating more than one gene, as well as more than one GO term. Therefore, we show in the last column of Table 2 the counts of evidence text passages in three different ways. The first number shows the total number of text passages with respect to (w.r.t) GO annotations: over 5500 text passages were used in the annotation of 1356 unique GO terms. So on average, each GO term is associated with four different evidence text passages in our corpus. The second number (5393) shows the total number of text passages with respect to different genes: for each of the 681 unique genes in our corpus, there are ∼7.9 associated text passages. Finally, the last number is the total number of unique text passages annotated in our corpus regardless of their association to either gene or GO terms.
Overall statistics of the annotated corpus grouped by data sets
| Data set | Articles | Genes (unique) | GO terms (unique) | Evidence text passagesw.r.t. GO|Gene|Unique |
|---|---|---|---|---|
| Training set | 100 | 316 | 611 | 2440|2478|1858 |
| Development set | 50 | 171 | 367 | 1302|1238|964 |
| Test set | 50 | 194 | 378 | 1763|1677|1253 |
| Total | 200 | 681 | 1356 | 5505|5393|4075 |
From Table 2 , we calculated that the average number of genes annotated in each article is 3.4, and the average number of GO terms associated with each gene is 2.0 in our corpus. Furthermore, as mentioned before, we have annotated two types of evidence text, depending on whether they contain experimental information. Accordingly, the two kinds are distinguished in our annotations by the presence or absence of associated evidence code. For the total 4075 unique pieces of evidence text, the majority (∼70%) of them contain experimental evidence. When broken down by databases, we see in Table 3 that results of FlyBase, MaizeGDB and TAIR are closer to the average statistics, while RGD and WormBase show some noticeable differences. Multiple factors can account for such differences including species, individual articles, curators and database curation guidelines.
Overall statistics of the annotated corpus grouped by MODs
| MOD | Articles | Genes (unique) | GO terms (unique) | Evidence text passagesw.r.t. GO|Gene|Unique |
|---|---|---|---|---|
| FlyBase | 39 | 140 | 267 | 1106|1106|881 |
| MaizeGDB | 30 | 85 | 193 | 664|595|492 |
| RGD | 88 | 236 | 369 | 1199|1223|946 |
| TAIR | 21 | 63 | 125 | 453|544|379 |
| WormBase | 22 | 157 | 402 | 2083|1925|1377 |
The location of evidence text and GO terms in the paper
Figure 3 shows the proportion of all evidence text in different parts of the article. As expected, the most informative location for extracting GO evidence text is the Results section, followed by the Discussion Section. Some GO evidence text also appears in Table or Figure legends. Within the full-text article, the Introduction/Background and Methods sections contain the least amount of information for complete GO annotation. Figure 3 also shows the limitation of using article abstracts for GO annotation: only 11.46% of the annotated text is found in the Title and Abstract combined. Of these, the majority (68.1%) were classified as summary sentences, while only 31.9% were experimentally supported sentences.
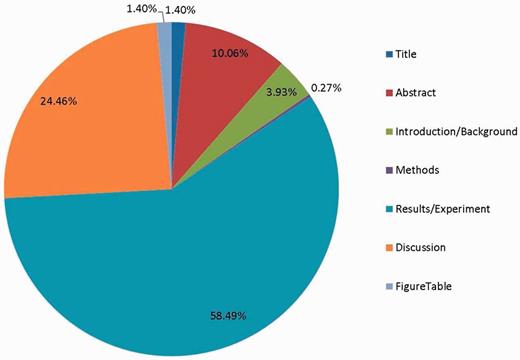
The proportion of annotated evidence text in different parts of the article.
Figure 4 shows the percentage of 1356 unique GO terms mentioned in different parts of the paper. Because a GO term might be mentioned in multiple locations, the sum of all percentages is greater than one in Figure 4 . As shown in Figures 3 and 4 , given 10% of relevant sentences in the abstract, one might identify more than 30% of the GO terms. Meanwhile, the Results/Experiment section remains the most information-rich location for mining GO terms.
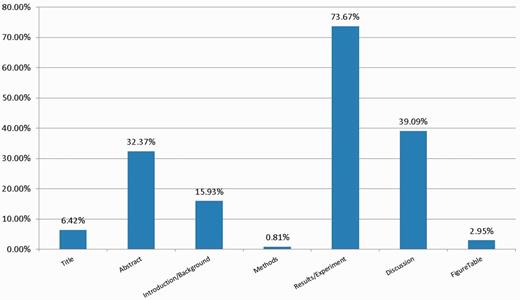
The proportion of GO terms appearing in different parts of the article.
IAA results
For evidence sentence selection, the IAA results are 9.3% (strict) and 42.7% (relaxed) in F 1 -measures, respectively. For GO term selection, the IAA results are 47% (strict) and 62.9% (hierarchical) in F 1 -measures, respectively. Our IAA result for the GO term selection (47%) is largely consistent with (also slightly better than) the previously reported 39% ( 45 ). Instead, our IAAs are more akin to the results found in a similar annotation task, known as MeSH indexing (IAA of 48%), in which human curators choose relevant annotation concepts from a large set of controlled vocabulary terms ( 46 , 47 ).
To better understand the discrepancies between annotators, we asked them to review the different annotations and reach a consensus. Furthermore, we separately characterized the source for those differences in both evidence sentence and GO term selection. For sentence selection, it is mostly due to missing annotations by one of the two annotators (76.6%), followed by selecting incomplete or incorrect sentences. Discrepancies in GO term selection are due to either missing (78.4%) or incorrect annotations (21.6%) where ∼23% of the latter can be counted as partial errors because annotated terms essentially differ in granularity (e.g. ‘response to fatty acid’ vs. ‘cellular response to fatty acid’). Finally, annotators do not always seem to agree on the set of genes for GO annotations in a given paper (IAA for gene selection is only 69%).
Conclusions and future work
Through collaboration with professional GO curators from five different MODs, we created the BC4GO corpus for the development and evaluation of automated methods for identifying GO terms from full-text articles in BioCreative IV ( 48 ).
There are some limitations related to this corpus that are worth mentioning. First, although the set of 200 papers in the BC4GO corpus is a good start for developing automated methods and tools, it is likely not enough, and the number of papers will need to be increased. As the ontologies and annotation methods of GO are continually expanding and improving, we feel that the training corpus will also need to continually expand and improve.
To ensure the positive and negative sentences are as distinct as possible, we asked our annotators to mark up every occurrence of GO evidence text. As a result, it greatly increased the annotation workload for each individual annotator. Given this time-consuming step, we chose to assign one annotator per article to maximize the number of annotated articles. In other words, our articles are not double annotated. Nonetheless, to assess the quality of our annotation as well as having a standard to compare with computer performance, we conducted a post-challenge IAA analysis by re-annotating 20 papers in the training set. Although we agree that IAA is important, we did not attempt to address IAA across the MODs in this work. One important consideration for IAA studies is that curators from different MODs have different expertise (e.g. plant biology vs. mammalian biology), and those differences can make it difficult for curators to confidently annotate papers outside of their area of expertise.
In addition, despite all our best efforts in ensuring consistent annotations (e.g. creating annotation guidelines, and providing annotator training), there will always be variation in the depth of annotation between curators and organisms as demonstrated in the post-challenge IAA analysis. For instance, there may be gray areas where some curators will select a sentence relating to a phenotype as a GO evidence sentence, while others will not. This result reflects the inherent challenge of GO curation as well as slight differences in annotation practice among the MODs.
Nonetheless, our work supports the idea that there is a great need for tools and algorithms to assist curators in adequately assigning GO terms at the correct level, especially as GO continues growing and more granular terms are added. We note, too, that our work provides additional evidence to support the assertion that redundancy of information within research articles allows for some leniency in evidence sentence recall ( 14 ). Such leniency should encourage developers of tools and algorithms in that text-mining applications do not need high sentence recall to achieve correspondingly high annotation recall ( 49 ). In the future, we plan to further assess the IAA for the complete corpus, for the sake of the improvement of those tools and algorithms.
The resulting BC4GO corpus is large scale and the only one of its kind. We expect our BC4GO corpus to become a valuable resource for the BioNLP research community. We hope to see improved performance and accuracy of text mining for GO terms through the use of our annotated corpus in the BC4GO task and beyond.
Acknowledgments
We would like to thank Don Comeau, Rezarta Dogan and John Wilbur for general discussion and technical assistance in using BioC, and in particular to Don Comeau for providing us source PMC articles in the BioC XML format. We also thank Lynette Hirschman, Cathy Wu, Kevin Cohen, Martin Krallinger and Thomas Wiegers from the BioCreative IV organizing committee for their support, and Judith Blake, Andrew Chatr-aryamontri, Sherri Matis, Fiona McCarthy, Sandra Orchard and Phoebe Roberts from the BioCreative IV User Advisory Group for their helpful discussions.
Funding
Intramural Research Program of the NIH, National Library of Medicine (to C.W., Y.M. and Z.L.), the USDA ARS (to M.L.S.), the National Human Genome Research Institute at the US National Institutes of Health (# HG004090, # HG002223 and # HG002273) and National Science Foundation (ABI-1062520, ABI-1147029 and DBI-0850319).
Conflict of interest . None declared.
References
Author notes
Citation details: Van Auken,K., Schaeffer,M.L., McQuilton,P. et al . BC4GO: a full-text corpus for the BioCreative IV GO task. Database (2014) Vol. 2014: article ID bau074; doi:10.1093/database/bau074

{kind=link}
{kind=link}
{kind=link}
{kind=link}