





Abstract
Renal neuroendocrine neoplasms are exceptionally rare and may mimic renal cell carcinoma on imaging. A 48-year-old man presented with one week of left flank pain. Ultrasound showed bilateral renal masses with a left subcapsular hematoma. Computed tomography and magnetic resonance imaging demonstrated multiple solid lesions in both kidneys with marked diffusion restriction, accompanied by a pancreatic uncinate mass, a right adrenal lesion, renal hilar lymphadenopathy, and a right retroperitoneal nodule. The patient underwent left nephrectomy and pancreatic biopsy. Histopathology confirmed neuroendocrine carcinoma involving the kidney and pancreas with nodal metastasis. This unusual constellation should prompt consideration of an underlying tumor predisposition syndrome such as von Hippel–Lindau disease, with recommendation for genetic counseling/testing and multidisciplinary surveillance. The patient improved after post-biopsy pancreatitis with conservative management and was referred for multidisciplinary oncologic management and imaging follow-up.
Introduction
Neuroendocrine neoplasms (NENs) most commonly arise in the gastrointestinal tract and bronchopulmonary system; primary renal NENs are exceptionally rare [1, 2]. In the current WHO/IARC framework, NENs span well-differentiated neuroendocrine tumors and poorly differentiated neuroendocrine carcinomas (NECs) [3]. Because neuroendocrine cells are not normally present in renal parenchyma, these tumors are difficult to diagnose preoperatively and may resemble more common renal malignancies on imaging [1, 2].
When multifocal bilateral renal masses coexist with pancreatic and adrenal lesions, a hereditary tumor predisposition syndrome such as von Hippel–Lindau (VHL) disease should be considered; expert guidance emphasizes genetic confirmation and structured lifelong surveillance of at-risk organs [4–6]. We report a rare constellation of bilateral multifocal renal neuroendocrine carcinoma with synchronous pancreatic neuroendocrine carcinoma and an adrenal lesion, and highlight key imaging findings and implications for syndromic evaluation.
Case report
A 48-year-old man was admitted with one week of persistent, dull left flank pain. He had no known personal or family history of hereditary tumor syndromes. Physical examination was unremarkable. Urinalysis showed no hematuria. Hemoglobin was 13.2 g/dL and white blood cell count was 13.36 K/μL with neutrophils 76.4%. Coagulation profile and hepatic/renal function were within normal limits.
Abdominal ultrasound demonstrated bilateral renal masses and a left subcapsular renal hematoma (Fig. 1). Multiphasic contrast-enhanced computed tomography (CT) revealed multiple bilateral renal tumors with a left subcapsular hematoma, bilateral renal hilar lymphadenopathy, a right retroperitoneal nodule, a mass in the pancreatic uncinate process, and a right adrenal lesion (Fig. 2).
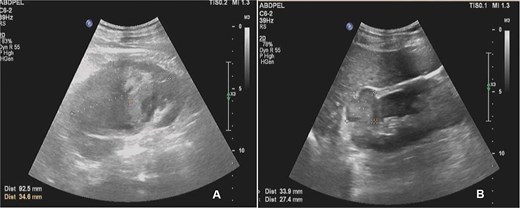
Abdominal ultrasound showing multiple solid masses in both kidneys and a subcapsular hematoma in the left kidney.
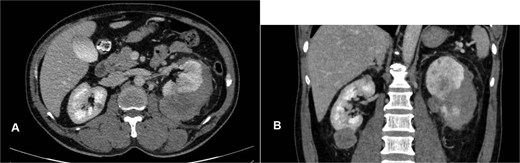
Contrast-enhanced CT showing multiple bilateral renal tumors, a left subcapsular hematoma, a pancreatic uncinate process mass, a right adrenal mass, a right retroperitoneal nodule, and bilateral renal hilum lymphadenopathy.
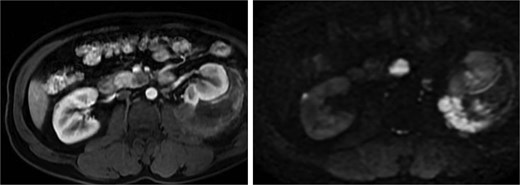
Abdominal magnetic resonance imaging (MRI) showed approximately seven to eight right renal masses (largest 32 × 35 × 37 mm) and five to six left renal masses (largest 62 × 55 × 55 mm). The renal lesions were iso- to mildly hypointense on T1-weighted imaging and mildly hypointense on T2-weighted imaging, without macroscopic fat. Post-gadolinium images demonstrated heterogeneous enhancement with central necrosis in the dominant masses (Fig. 3). Diffusion-weighted imaging showed marked restriction in the viable tumor components (Fig. 4). The dominant left renal mass, located in the mid kidney, demonstrated invasion of the posterior renal fascia. Subacute hemorrhage under the left renal capsule measured up to 24 mm in thickness.
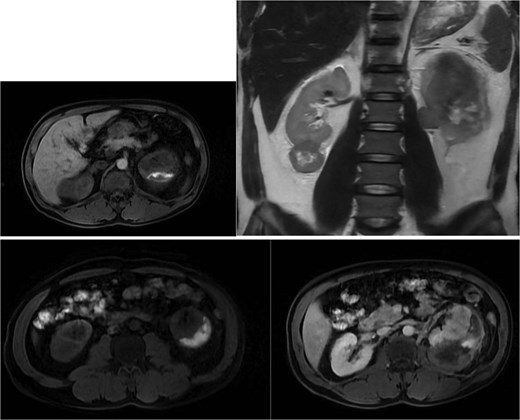
Axial T1-weighted and T2-weighted MRI showing multiple solid renal masses with low signal intensity and a large left subcapsular hematoma.
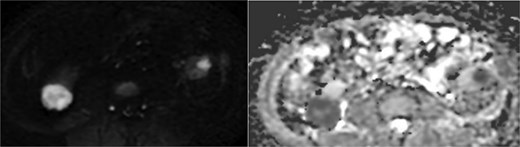
Diffusion-weighted imaging (b = 800) and ADC maps showing restricted diffusion in the viable components of bilateral renal tumors.
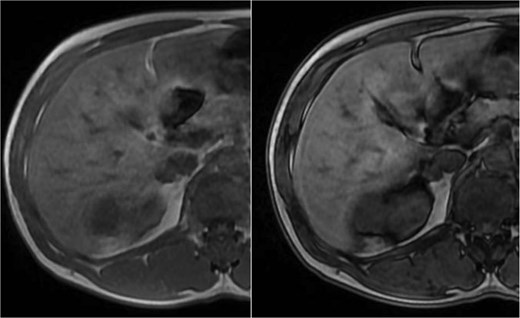
A pancreatic uncinate process mass measured 18 × 20 mm and was hypointense on T1-weighted and mildly hyperintense on T2-weighted imaging, with relative hypoenhancement compared with adjacent pancreatic parenchyma and marked diffusion restriction (Fig. 5). A right adrenal mass measured 12 × 16 mm, was mildly hyperintense on T2-weighted imaging, and showed contrast enhancement and diffusion restriction (Fig. 6). Brain MRI demonstrated no intracranial lesions.
MRI of the pancreatic uncinate process mass showing low T1 signal, mildly increased T2 signal, restricted diffusion, and relative hypoenhancement on post-contrast images.
MRI of the right adrenal lesion showing mild T2 hyperintensity, contrast enhancement, and restricted diffusion.
The patient underwent left nephrectomy and biopsy of the pancreatic lesion. Histopathology demonstrated neuroendocrine carcinoma involving the kidney and pancreas, with lymph node metastasis (Fig. 7). Acute pancreatitis occurred after pancreatic biopsy and improved with conservative management.
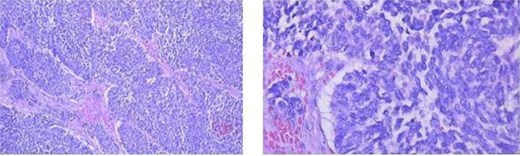
Histopathology showing neuroendocrine carcinoma in the kidney and pancreas (hematoxylin–eosin stain) and lymph node metastasis.
The patient recovered and was discharged after 7 days. The case was reviewed in a multidisciplinary tumor board, and further management was planned with medical oncology, including consideration of systemic therapy and imaging surveillance of the contralateral renal lesions, adrenal lesion, and retroperitoneal nodule. Given the constellation of renal, pancreatic and adrenal lesions, genetic counseling/testing for VHL disease was recommended, and endocrine evaluation of the adrenal lesion (including biochemical testing for pheochromocytoma/paraganglioma) was advised because pheochromocytoma can be clinically silent. At the 3-month follow-up, the patient remained clinically stable, and surveillance imaging showed no interval progression of the contralateral renal lesions, adrenal lesion, or retroperitoneal nodule.
Discussion
Primary renal NENs are exceptionally rare, and most evidence is derived from case reports and small series [1, 2, 7]. Because imaging findings are heterogeneous and may overlap with more common renal entities, histologic confirmation is often required, particularly in bilateral or multifocal presentations.
The radiologic differential diagnosis for multifocal bilateral renal masses includes multifocal or hereditary renal cell carcinoma, renal lymphoma, and metastases. Marked diffusion restriction is not specific and can also be seen in lymphoma and in some hypovascular renal carcinomas. In our patient, the concomitant pancreatic and adrenal lesions increased suspicion for a syndromic context.
Primary site consideration is challenging in this case. The dominant renal mass was the symptomatic lesion and demonstrated locally aggressive behavior (posterior renal fascia invasion) with multifocal bilateral renal involvement and renal hilar nodal disease on imaging, which favors the kidney as a clinically dominant site. However, because synchronous renal and pancreatic neuroendocrine carcinoma is exceedingly rare and histologic/molecular comparison between sites was limited, the primary site cannot be definitively assigned. We therefore describe this presentation as synchronous renal and pancreatic NEC involvement, which is a limitation of the case.
VHL disease is a multisystem tumor predisposition syndrome that can involve the kidneys, pancreas and adrenal glands. International and expert-consensus recommendations emphasize genetic confirmation and structured, lifelong surveillance of at-risk organs [4–6]. Radiology is central to surveillance of renal manifestations in VHL [8]. Although VHL-associated renal tumors are classically clear cell renal cell carcinoma rather than neuroendocrine carcinoma, the phenotype in this case warrants genetic counseling/testing to confirm or exclude VHL and to guide screening and counseling of at-risk relatives.
Management of renal neuroendocrine carcinoma is not standardized and is generally extrapolated from neuroendocrine carcinoma at other sites. Surgical resection is preferred for localized disease when feasible; systemic therapy (often platinum-based) is commonly considered for advanced poorly differentiated disease, although outcomes are variable [7, 9]. If VHL is confirmed, multidisciplinary management is required for the spectrum of potential manifestations. Targeted systemic options such as the HIF-2α inhibitor belzutifan may reduce the need for repeated interventions in selected VHL-associated neoplasms [10]. Because pheochromocytoma can be clinically silent in VHL, adrenal lesions should be phenotyped with appropriate biochemical testing [11].
VHL-associated pancreatic neuroendocrine tumors are often nonfunctional but carry metastatic risk. Tumor size, growth kinetics and genotype have been linked to metastatic potential, supporting risk-adapted surveillance and management strategies [12–15].
Conclusion
Bilateral multifocal renal neuroendocrine carcinoma with synchronous pancreatic neuroendocrine carcinoma and an adrenal lesion is exceptionally rare. When CT/MRI demonstrates multifocal bilateral renal masses accompanied by pancreatic and adrenal tumors, clinicians should consider a syndromic association such as VHL and recommend genetic counseling/testing and multidisciplinary surveillance.
Conflicts of interest
The authors declare that they have no competing interests.
Funding
None declared.
Patient consent
Written informed consent was obtained from the patient for publication of this case report and accompanying images. A copy of the written consent is available for review by the Editor-in-Chief on request.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}