





Abstract
Extracranial metastases from meningioma are exceedingly rare, representing <1% of all cases, with appendicular skeletal involvement being exceptional. We report a 51-year-old woman who developed a destructive iliac bone lesion ten years after complete resection of a WHO Grade II (atypical) parietal meningioma. Computed-tomography-guided biopsy confirmed metastatic meningioma morphologically identical to the original tumor. A systematic review (1959–2024) identified 26 reported cases of appendicular skeletal metastases, most frequently involving the femur and iliac bone, with a mean latency of 8.9 years. These findings emphasize the indolent yet unpredictable metastatic potential of atypical meningiomas and highlight the importance of lifelong surveillance, as metastasis should be considered even after prolonged disease-free intervals.
Introduction
Meningiomas constitute approximately 37% of all primary central nervous system (CNS) tumors and are graded by the World Health Organization (WHO) as Grade I, II, or III according to histopathologic criteria [1, 2]. While the majority are benign, approximately 10%–15% exhibit atypical or anaplastic features associated with recurrence or dissemination. Extracranial metastases occur in <1% of cases, with preferential spread to the lungs, liver, lymph nodes, and bone [3, 4]. Osseous metastases usually affect the axial skeleton; appendicular involvement accounts for <15% of reported cases [5, 6]. Diagnosis can be challenging, as radiologic appearances often mimic metastatic carcinoma or primary bone sarcoma. Confirmation relies on histopathologic correlation and immunohistochemical (IHC) positivity for epithelial membrane antigen (EMA), vimentin, and progesterone receptor (PR) with cytokeratin negativity [4, 7]. We report a rare case of iliac metastasis arising a decade after resection of a Grade II meningioma, accompanied by a review of the literature on appendicular skeletal involvement.
Case presentation
A 51-year-old woman presented in 2014 with headache and progressive right-sided weakness. MRI showed a lobulated right parietal extra-axial mass (7 × 5 × 5 cm) with homogeneous enhancement, vasogenic oedema, and midline shift. She underwent complete surgical resection followed by adjuvant radiotherapy. Histopathology demonstrated spindle-cell and meningothelial proliferation arranged in whorls with necrosis and a mitotic count of 6/10 HPF (Fig. 1). IHC was positive for EMA and vimentin but negative for cytokeratin, consistent with atypical meningioma (WHO Grade II). Ten years later, she developed progressive left-hip pain. Computed tomography (CT) of the pelvis revealed a lytic destructive lesion involving the left iliac bone with soft-tissue extension (Fig. 2). CT-guided biopsy showed tumor cells morphologically identical to the original specimen. IHC was positive for EMA and PR and negative for S-100, confirming metastatic meningioma (Fig. 3). Systemic imaging showed no visceral metastases.
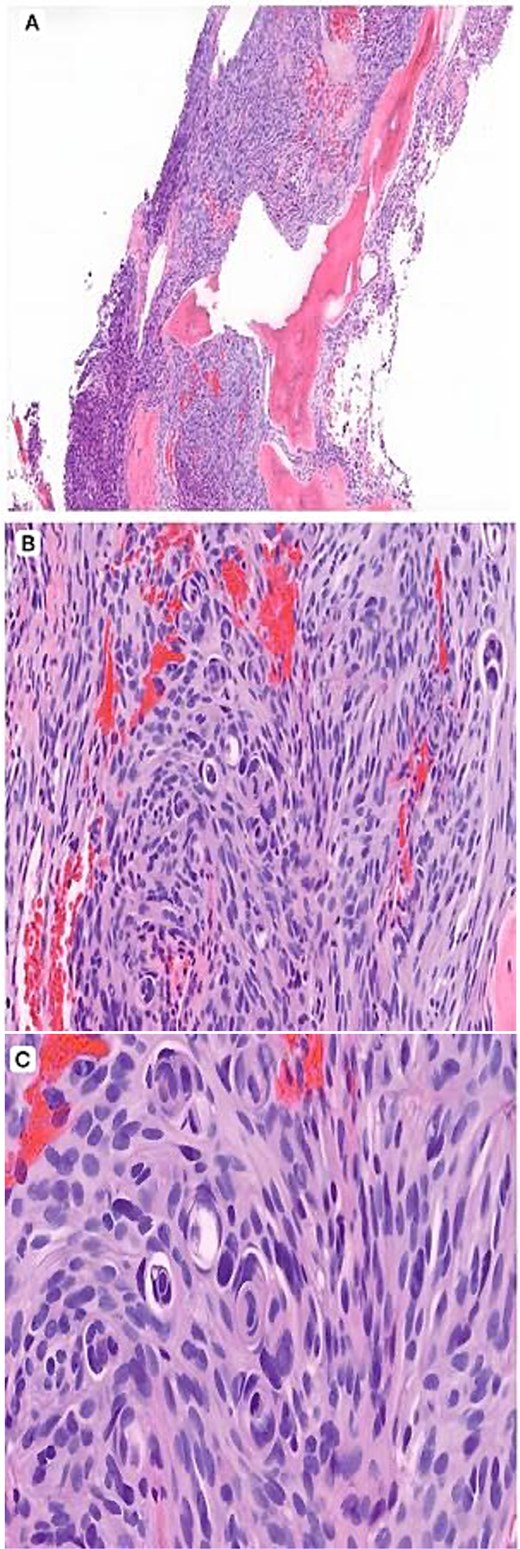
Histopathology (H&E). (A) Low-power view (×4) showing a spindle cell neoplasm infiltrating lamellar bony trabeculae. (B) Intermediate-power view (×10) demonstrating meningothelial proliferation arranged in lobules with early whorl formation. (C) High-power view (×20) showing well-formed whorls and characteristic intranuclear pseudoinclusions, supporting a meningothelial neoplasm.
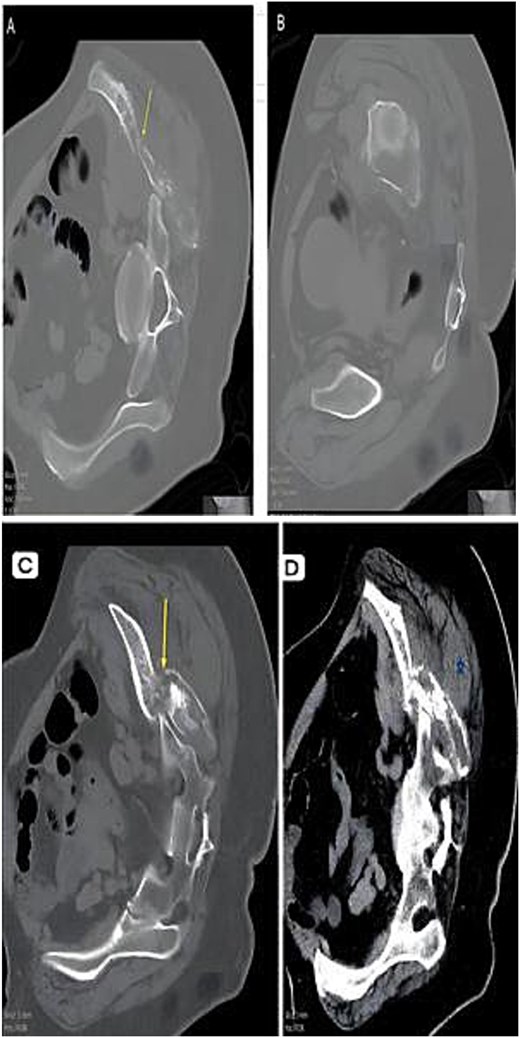
CT composite. (A) Axial CT—Lytic destructive lesion with cortical breach. (B) Follow-up CT—Progressive osseous destruction & soft-tissue expansion. (C) Bone window—Cortical destruction (arrow). (D) Contrast CT—Enhancing soft-tissue mass replacing marrow (asterisk).
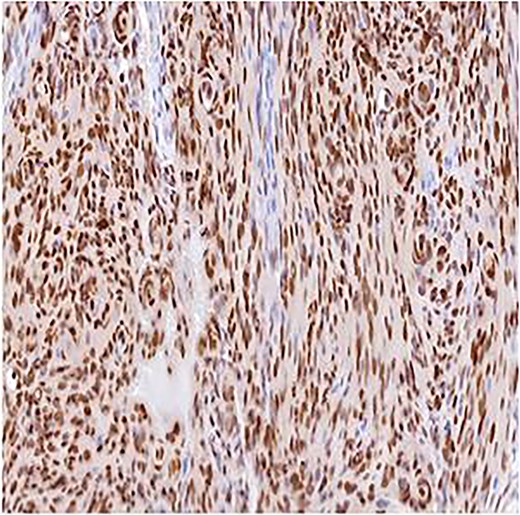
Immunohistochemistry for PR, ×10. Strong nuclear staining within meningothelial tumor cells confirming meningioma differentiation.
The patient had a single metastatic focus, and comprehensive evaluation revealed no additional disease elsewhere in the body. Given the absence of symptoms and the complete excision of the lesion, a decision was made to proceed with close clinical and radiological observation rather than adjuvant therapy. Over the subsequent 20 months of follow-up, she has remained clinically well and radiologically stable, with no evidence of recurrence.
Discussion
Appendicular metastasis from atypical meningioma is exceedingly rare but clinically significant. The ten-year latency in this case highlights the unpredictable nature of meningioma Metastatic meningioma remains a diagnostic and therapeutic challenge due to its rarity and often indolent course. Appendicular skeletal metastases are exceptionally uncommon but increasingly reported in atypical and anaplastic subtypes [4, 5]. A systematic MEDLINE search (1959–2024) identified 26 histologically confirmed cases of appendicular skeletal metastasis from meningioma. The mean age at metastatic presentation was 49 years (range 27–75) with a nearly equal sex distribution, contrasting with the known female predominance in intracranial disease [8]. Most appendicular metastases arose from WHO Grade II (44%) or III (26%) tumors, while Grade I accounted for 30% after long latency periods. The mean interval from initial craniotomy to metastasis was 8.9 years (range 2–27). The femur was most frequently involved (52%), followed by the iliac bone (20%), humerus (16%), and clavicle (8%), likely reflecting haematogenous spread through Batson’s valveless venous plexus [9]. Patients typically presented with localized pain or pathological fracture. Imaging revealed lytic or mixed lytic–sclerotic lesions with cortical destruction. Despite the rarity, survival exceeded 50% at five years for those with solitary appendicular lesions, indicating that tumor burden and metastatic site may be more prognostically significant than grade alone. The overall mortality rate was 41%, but patients with solitary appendicular metastases demonstrated >50% five-year survival, suggesting that tumor burden and metastatic site may be more prognostically relevant than histologic grade alone.
The mechanism of spread is primarily haematogenous via the dural venous sinuses or retrograde venous flow [9]. Histologically, metastatic deposits recapitulate the meningothelial morphology of the primary tumor. PR expression supports a low-to-intermediate-grade lesion, whereas PR loss, high mitotic activity, and NF2 or TERT promoter mutations are linked with aggressive or metastatic potential [10, 11]. Recent genomic analyses have identified driver alterations in NF2, TERT, PI3K/AKT/mTOR, and SMO/TRAF7 pathways that underlie progression and recurrence [12, 13]. Chromosomal instability, particularly 1p/14q co-deletion and CDKN2A/B loss, correlates with adverse outcome [14]. These discoveries have prompted exploration of targeted and immune-based therapies in advanced disease. Surgery and adjuvant radiotherapy remain mainstays for local control, but systemic treatment is required for disseminated or unresectable lesions. VEGF pathway inhibitors such as bevacizumab have shown radiographic stabilization and symptomatic relief [13]. mTOR inhibitors such as everolimus, alone or combined with somatostatin analogs, achieve disease stabilization by targeting the PI3K/AKT/mTOR axis [14]. Immune checkpoint inhibitors (nivolumab, pembrolizumab) demonstrate modest benefit in PD-L1–expressing or hypermutated tumors [15]. Peptide receptor radionuclide therapy (PRRT) with 177Lu-DOTATATE offers another experimental option for refractory disease [13]. Translational approaches including circulating tumor DNA (ctDNA) and radiogenomic profiling may refine prognostication and allow personalized therapy [12].
Awareness of this phenomenon, combined with molecular understanding and emerging targeted therapies, will improve recognition and management of this uncommon manifestation.
Conflict of interest statement
The authors declare that they have no conflicts of interest.
Funding
None declared.

{kind=link}
{kind=link}
{kind=link}