





Abstract
A solitary fibrous tumor (SFT) is a rare neoplasm with a low metastatic rate. We report a case of a SFT with bone and pulmonary dissemination of an unusual presentation. A 69-year-old male presented with progressive rib pain of 2 years of evolution, which migrated to the ipsilateral lower extremity, progressively compromising its mobility. The diagnosis of SFT was confirmed by immunohistochemical analysis, and the presence of metastasis was suggested by imaging findings and confirmed by histopathological examination. This case shows an uncommon neoplasm with an atypical clinical presentation in terms of age, stage, and tumor behavior. Despite initial palliative measures and radiotherapy, the patient experienced worsening symptoms, including severe pain, limb shortening, scrotal edema, and anasarca. Chemotherapy with ifosfamide and doxorubicin was initiated but had to be discontinued due to cardiotoxicity and clinical decline, ultimately leading to the patient’s death within less than a year from presentation.
Introduction
Chest pain accounts for 1% of all outpatient visits in primary care [1]. Its evaluation represents a diagnostic challenge due to the wide range of possible etiologies. Common causes include chest wall pain, reflux esophagitis, and costochondritis [2]. However, there are less common etiologies that can make diagnosis difficult.
A solitary fibrous tumor (SFT) is a rare mesenchymal neoplasm characterized by a proliferation of fibroblastic cells. Although it usually originates in the pleura, it can also occur in other extrapleural locations. It represents ~3.7% of soft tissue sarcomas and mesenchymal tumors of intermediate malignancy [3]. Its annual incidence is one new case per 1 million people [4]. It can manifest itself at any age, although it is most frequently observed in the fifth and sixth decades of life [5]. There is no gender predisposition, and no associated hereditary or environmental risk factors have been identified [6].
The symptomatology of SFT usually varies according to the site of presentation and the tumor size, and it can be asymptomatic and detected incidentally through imaging studies performed for other causes [7]. Its diagnosis is based on compatible imaging findings, although definitive confirmation requires histopathological and immunohistochemical analysis [5, 7]. Standard treatment includes surgery, with radiotherapy and chemotherapy options in selected cases [5].
We present the case of a male patient who presented pain in the right costal region that progressively increased and migrated to the leg on the same side, causing limitation of movement, and was subsequently diagnosed with a metastatic SFT.
Case report
A 69-year-old male patient from Ferreñafe, Peru, attended the internal medicine outpatient clinic of the Regional Hospital of Lambayeque in October 2023. He reported mild and occasional pain in the right costal region since May 2022, which initially subsided with nonsteroidal anti-inflammatory drugs (NSAIDs). However, in December of the same year, the pain intensified, becoming persistent and associated with discomfort in the right leg that progressively caused difficulty walking.
During the physical examination, a decrease in the range of motion of the right hip and mechanical pain were observed, with no other relevant signs or symptoms. Given these findings, radiological studies were requested. In his personal history, he reported no relevant comorbidities; however, he mentioned that his sister had been diagnosed with lung cancer.
The pelvic radiograph showed a lytic lesion measuring 8 × 8 cm on the outer edge of the right iliac bone (Fig. 1), which was initially considered as a giant cell tumor. Given these findings, a computed tomography (CT) scan of the thorax, abdomen, and pelvis was requested to evaluate possible tumor dissemination. The CT scan revealed a heterogeneous expansive mass measuring 113 × 96 × 100 mm with destruction and remodelation of the right iliac bone (Fig. 2), a 45 × 38 mm mass in the right chest wall with heterogeneous contrast (Fig. 3), three pulmonary nodules in the left lower lobe (5–12 mm) (Fig. 4A) and multiple hypo- and isodense hepatic lesions (10–28 mm in both hepatic lobes) (Fig. 4B). That same month, two biopsies were performed. The first, from the right chest lesion, showed positivity for CD34, BCL-2, and STAT6 in the immunohistochemical study, confirming the presence of an SFT. The second, from the right iliac crest lesion, showed a spindle-cell sarcoma with bone invasion, with immunohistochemistry positive for CD34 and vimentin, a Ki-67 index of 10%, and negative for cytokeratins. This biopsy was processed in a public hospital pathology laboratory, where reagents for BCL-2 and STAT6 were not available at the time of analysis. Based on the histologic appearance, partial immunophenotype, and clinical context, the iliac lesion was considered highly suggestive of metastasis from the chest wall tumor. However, due to the patient’s rapid clinical deterioration and subsequent death, confirmatory testing by STAT6 immunostaining could not be performed.
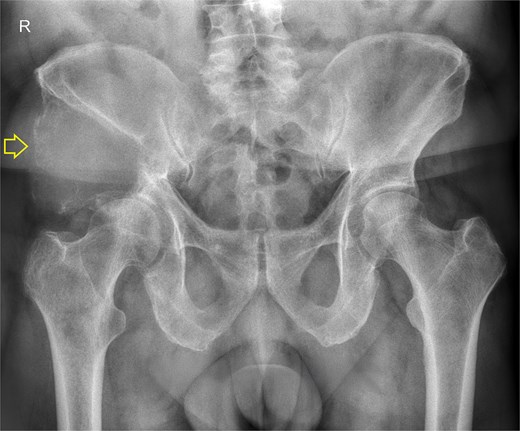
Pelvis radiograph, anterior view. A well-defined lytic lesion measuring 8 × 8 cm is identified in the right iliac wing.
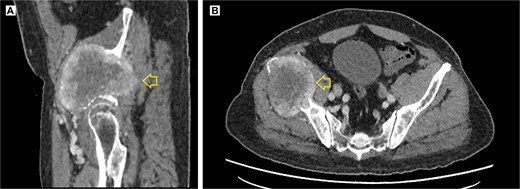
Contrast-enhanced pelvic CT scan. (A) Sagittal view of tumor in iliac crest. (B) Axial view of the iliac crest. Expansile soft tissue mass (arrow) of 113 × 96 × 100 mm, with central necrotic degeneration and heterogeneous contrast enhancement but predominantly peripheral.
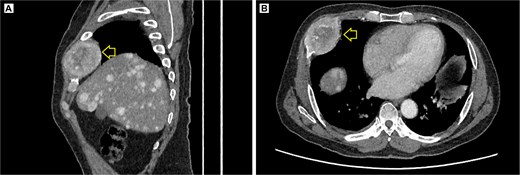
Contrast-enhanced thoracic CT scan. (A) Sagittal view showing the tumor on the chest wall, involving a rib. (B) Axial view showing the tumor in the right costal arch. Expansile soft-tissue mass (arrow) measuring 45 × 38 mm in the right costal arch, with central necrotic degeneration and heterogeneous peripheral contrast enhancement.
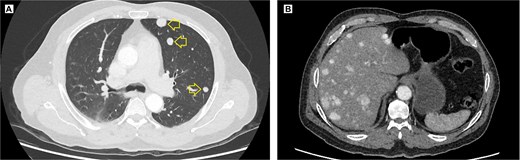
Contrast-enhanced thoracic and abdominal CT scan. (A) Axial view in the lung window of the thorax, solid hyperenhancing parenchymal nodules (arrow) ranging from 5 to 12 mm are observed in the left lung. (B) Coronal view in the soft tissue window, solid hyperenhancing nodules measuring 10 to 28 mm with diffuse distribution are observed.
In January 2024, the patient attended the oncology department reporting severe and persistent pain in the right leg (7/10 on the visual analog scale), limiting his ability to ambulate. He also described sharp pain in the right hemithorax, occasionally radiating to the back and right hip. Palliative treatment with tramadol and naproxen twice a day was started, achieving partial relief. Physical examination revealed a palpable tumor in the posterolateral costal wall and a shortening of the right lower limb of ~1 cm.
In March 2024, he started radiotherapy, completing 10 sessions. During a medical consultation 2 months later, he presented with severe scrotal edema, and anasarca, with a weight gain from 82 to 102 kg. A soft tissue ultrasound of the left arm was requested, and a solid mass was found at the level of the muscular plane, with a diagnostic possibility of cancerous dissemination. The patient started chemotherapeutic treatment in May 2024 with ifosfamide (4100 mg) and doxorubicin (27 mg); however, only three cycles were completed due to the development of anthracycline-induced heart disease and progressive deterioration of his general clinical condition. The patient ultimately died on 3 October 2024. A timeline summarizing the key clinical events, diagnostic procedures, treatments, and outcomes is shown in Fig. 5.
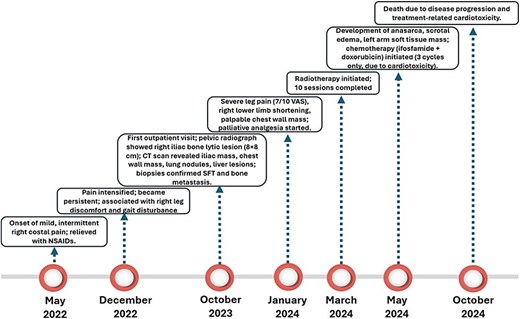
Timeline of case report.
Discussion
An SFT is a rare neoplasm that usually occurs in patients between the fifth and sixth decades, with a mean age of 50.30 years in extrapleural cases. Initial truncal localization accounts for ~10% of reported cases. Due to its asymptomatic course in early stages, its detection is usually incidental, and when symptoms occur, they are a consequence of compression of adjacent structures [5]. According to the 2002 World Health Organization classification, an SFT is considered a pathology with a low risk of metastasis [8].
The reported case corresponds to a 69-year-old patient, an advanced age compared to the average for the typical presentation. In addition, the tumor presented considerable size and necrosis area, features that have been identified in the literature as predictors of malignancy and metastasis [5, 8]. In a study by Demicco et al., tumors with metastatic behavior were associated with older patient age (median 61 vs. 51 years; P = .0043), larger tumor size (median 16.20 vs. 8.70 cm; P < .0001), and presence of necrosis (58.00% vs. 29.00%; P = .0170). Likewise, age and tumor size were correlated with higher mortality [8].
The diagnosis was made 19 months after the onset of symptoms, a diagnostic delay that could have been mitigated with a better anamnesis and physical examination at the primary care level, which would have allowed radiological tests to be performed in a timelier manner. This highlights the need to sensitize health professionals to the early identification and referral of rare neoplasms such as SFTs.
Definitive diagnosis of an SFT is based on immunohistochemistry, with markers such as CD34, CD99, and BCL-2 showing strong diffuse expression in ~90% of cases [5]. In this patient, CD34, STAT 6, and BCL-2 markers were positive, confirming the diagnosis. However, the inclusion of immunohistochemical expression of TP53 could have provided additional information, as this marker is associated with increased recurrence and metastasis [5]. Also, one study found an association between TP53 immunopositivity and SFT malignancy [9].
Certain histologic features suggestive of malignancy have been described in SFTs, such as increased cellularity, increased mitotic activity (≥4 mitoses per 10 high-power fields or >2 mitoses per 2 mm2), nuclear pleomorphism, tumor necrosis, and infiltrative borders [5]. These alterations are associated with more aggressive clinical behavior, a higher likelihood of recurrence and metastasis, and reduced overall survival [8, 10]. It should be noted that some tumors, even if they do not initially exhibit these features in the primary resection specimen, may develop them during tumor progression [5].
Although surgical resection is the standard treatment for localized SFTs, in this case, the presence of widespread metastatic disease, along with progressive clinical deterioration and poor functional status, rendered the patient ineligible for curative surgery. The extensive iliac bone involvement and the need for complex reconstruction further limited surgical feasibility. Therefore, management was focused on palliative measures aimed at symptom control and quality of life, including radiotherapy and systemic chemotherapy.
In the present case, histological signs of tumor aggressiveness were not reported, limiting the accuracy of prognostic assessment. However, since the diagnosis occurred at an advanced metastatic stage, a palliative treatment regimen including radiotherapy and chemotherapy with ifosfamide and doxorubicin was indicated, consistent with protocols used for aggressive sarcomas and soft tissue tumors [11]. While molecular studies provide precise diagnosis, their high cost and need for specialized personnel restrict their use in resource-limited settings, where immunohistochemistry remains the most practical, sensitive, and specific diagnostic tool for SFTs [5].
This case highlights the challenges of managing SFTs diagnosed at an advanced stage. The absence of histopathological indicators associated with malignancy, such as increased cellularity, mitotic activity, or infiltrative borders, does not preclude aggressive clinical behavior, as evidenced by distant metastases justifying a palliative approach. It underscores the importance of timely diagnosis and comprehensive histological characterization to guide appropriate follow-up and anticipate disease progression, particularly where molecular diagnostics are inaccessible.
Learning points
SFT are rare mesenchymal neoplasms with generally indolent behavior, but may present with aggressive features, including metastasis to bone and lung, especially in older patients.
Immunohistochemical markers such as CD34, BCL-2, and STAT6 remain crucial for diagnosis, particularly in resource-limited settings where molecular testing is unavailable.
Large tumor size, necrosis, and patient age are key prognostic indicators of malignant potential in SFTs, even in the absence of classical histologic features of malignancy.
Early detection and thorough initial evaluation are essential, as delays in diagnosis can lead to advanced disease and limit curative treatment options.
The presence of atypical presentations, such as progressive limb pain or soft tissue metastases, should prompt clinicians to consider rare neoplasms in the differential diagnosis, particularly when initial treatments fail.
Author contributions
K.A.Z.C. conceptualized the clinical case, managed permissions and resources for it, and reviewed the final version. A.Z.V.C. wrote the initial draft and reviewed the final version. J.E.S.F. conducted the research, drafted the initial draft, and drafted and reviewed the final version.
Conflict of interest statement
The authors declare that they have no conflicts of interest with respect to this article.
Funding
This article was self-funded by the authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}