





Abstract
Peutz-Jeghers syndrome (PJS) is an autosomal dominant mutation of the STK11/LKB1 gene on chromosome 19 often characterized by mucocutaneous pigmentation, hamartomatous polyps, anemia, gastrointestinal bleeding and intussusception. We present the case of a 21-year-old female with no pertinent family history who received the diagnosis of PJS after presenting to the hospital with two episodes intussusception. Patients with PJS have an increased lifetime risk of developing stomach, small bowel, colon, pancreatic, breast, cervical, uterus and testicular cancer requiring religious surveillance at an early age.
INTRODUCTION
Peutz-Jeghers syndrome (PJS) is an autosomal dominant mutation of the STK11/LKB1 gene on chromosome 19 often characterized by mucocutaneous pigmentation, hamartomatous polyps, anemia, gastrointestinal bleeding and intussusception. We report the case of a 21-year-old female who presented with acute onset of abdominal pain associated with nausea and emesis with evidence of intussusception of 15 cm of the small intestine on imaging, confirmed as PJS on pathology evaluation of the tissue specimen. PJS is a rare condition with risks of associated malignancy; early diagnosis with indicated intervention and appropriate regular follow up are imperative for positive prognosis in patients affected by the rare syndrome.
CASE REPORT
Patient is a 21-year-old female who presented with severe, sudden onset abdominal pain with associated poor oral intake, nausea and vomiting. Patient’s only past medical history was remarkable for normocytic anemia found on lab work. Patient did not report any significant family history. As a part of the patient’s initial work-up a computed tomography (CT) of the abdomen and pelvis with IV contrast was obtained with evidence of intussusception of a long segment of small bowel in the lower midline abdomen extending into the pelvis, measuring ~15 cm in length (Fig. 1); the patient’s CT was not remarkable for pneumatosis or obstruction.
The patient was subsequently taken to the operating room for diagnostic laparoscopy where the area of intussusception was identified; the case was then converted to open as the area of intussusception was too large to reduce laparoscopically. During the exploratory laparotomy, two independent sites of intussusception with associated masses were noted, manually reduced and resected. The specimen was sent to pathology, where multiple Peutz-Jeghers hamartomas were noted without evidence of dysplasia or malignancy. Numerous sessile and pedunculated polyps ranging from 0.2 to 3.7 cm in greatest dimensions were noted.
The patient developed abdominal pain with associated leukocytosis with left shift, which prompted a CT of the abdomen and pelvis on postop Day 4 with evidence of moderate to large amount of ascites and a large amount of scattered pneumoperitoneum (Fig. 2).
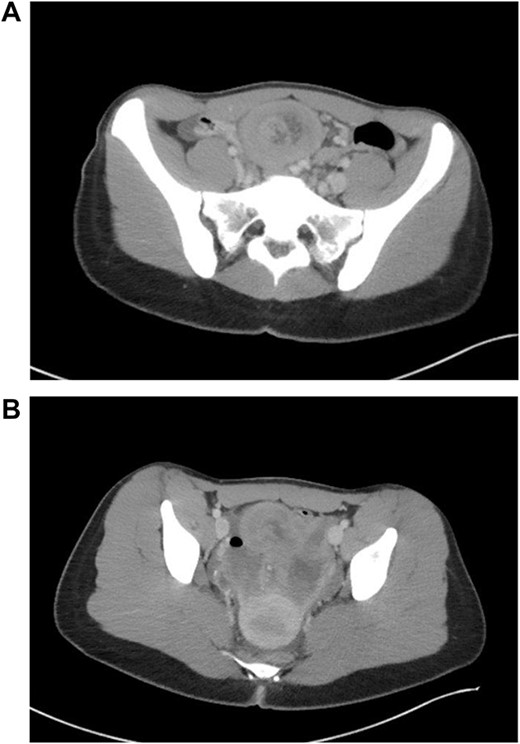
CT findings of long segment lead point.
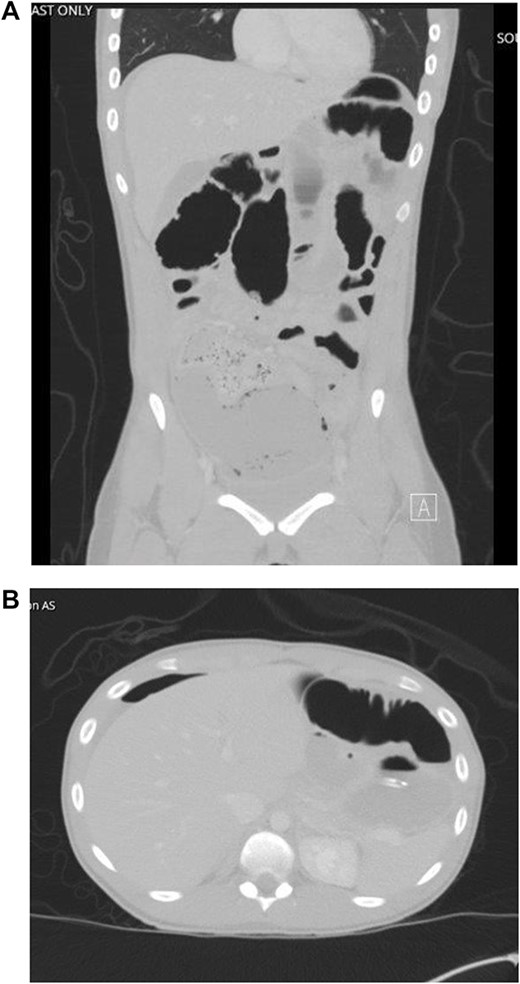
CT imaging with evidence of ascites and scattered pneumoperitoneum.
The decision was made on postop Day 4 to return to the operating room for a second exploratory laparotomy utilizing the previous incision, where a large amount of succus in the abdomen was identified. It was noted that a corner of the proximal anastomosis had a small leak with easy expression of succus. No new anastomosis was created at that site, and the specimen was sent for pathology with evidence of 12 sessile polyps measuring up to 0.3 cm.
The patient was tolerating a diet, and experiencing anterograde bowel function on postop Day 9; however, the decision was made to obtain a CT of the abdomen pelvis due to an increasing leukocytosis shift. CT revealed two fluid collections with gas pockets, anteriorly measuring 4.0 × 2.8 cm, 9.8 × 6.3 cm in the rectal space (Fig. 3).
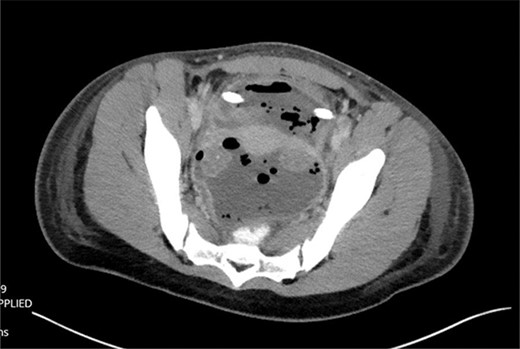
CT findings of pelvic fluid collection.
A drain was placed by interventional radiology, drain fluid growing Proteus mirabilis, Klebsiella pneumonia and yeast. After several days of observation, the patient was discharged on oral antibiotics with plans for interval removal of drain 2 weeks later.
DISCUSSION
PJS is a rare autosomal dominant disorder due to a mutation in the serine threonine kinase STK11/LKB1 gene on chromosome 19, which impacts 1/50 000–200 000 individuals. Diagnosis is considered in patients with (1) two or more Peutz-Jeghers polyps, (2) any number of polyps with family history of PJS, (3) mucocutaneous pigmentations with family history of PJS or (4) any number of Peutz-Jeghers polyps with mucocutaneous pigmentations [1]. The patient in our presentation had multiple Peutz-Jeghers polyps and mucocutaneous pigments on the lips and hands, meeting the inclusion criteria for the syndrome. Approximately 95% of patients with PSJ have mucucutaneous pigmentation, typically located around the mouth, eyes, nostrils, and less commonly on hands or feet [2].
PJS may manifest in both inherited or sporadic patterns, with significant clinical heterogeneity. The PJS gene is located on chromosome 19p34-p36, and codes for the serine–threonine kinase (STK11) a tumor suppressor gene; a mutation in this gene leads to uncontrolled cell growth, which results in the development of hamartomas and cancers [2, 3] Patients with PJS require multidisciplinary evaluation and surveillance due to an increased lifetime cancer risk of nearly 90% by the age of 70 [4, 5]. Patients have an elevated risk of gastrointestinal, breast, cervical, uterine, pancreas and lung cancer [6].
PJS patients’ care require a lifelong multidisciplinary approach towards cancer surveillance given significantly elevated cancer risk [7]. Current recommendations include annual hemoglobin concentration, esophagogastroduodenoscopy every 2–3 years, small bowel series/enteroscopy every 2 years without any consensus on initial age of surveillance. Colonoscopy or flexible sigmoidoscopy and barium enema every 2–3 years from onset of symptoms or at the age of 25. Annual abdominal ultrasonography, or MRCP/ERCP every 2–3 years from age of 25. At least annually breast exams at the age of 18, mammogram or magnetic resonance imaging (MRI) every 2 years, annually subsequent to age of 50. Annual pelvic exam, pelvic ultrasound and cervical smears; 7 sources recommend serial CA-125, and endometrial biopsy annually from the age of 20. For men, annual testicular exam, ultrasound if symptomatic from birth [3, 8–10] (Table 1).
| Cancer type | Age to start screening | Screening modality | Screening interval |
|---|---|---|---|
| Stomach | No consensus, recommendations for early screening | EGD | 2–3 |
| Small bowel | No consensus, recommendations for early screening | Capsule endoscopy | 2–3 |
| Colon | 25 | Colonoscopy | 2–3 |
| Pancreas | 25 | Ultrasound or MRCP/ERCP | Modality dependent: ultrasound – 1 year, MRCP/ERCP 2–3 |
| Breast | 18 | Breast Exam, MRI/mammography | Breast exam – annually MRI/Mammogram – 2 years |
| Cervical | 20 | Cervical smear | 1 |
| Uterus | 20 | Pelvic Ultrasound/pelvic exam | 1 |
| Testicular | Birth | Testicular exam/ultrasound | 1 |
CONCLUSION
PJS is a rare autosomal dominant disorder which impacts 1/50 000–20 000 individuals, with a significant lifetime risk of malignancy. We presented a rare case of a sporadic PJS, where the patient the patient and family denied any family history. Therefore, it is imperative for surgeon to be aware of the clinical manifestations, diagnostic criteria and surveillance recommendations of PSJ.
CONFLICT OF INTEREST STATEMENT
None declared.

{kind=link}
{kind=link}
{kind=link}