





Abstract
Hirschsprung disease (HSCR) is characterized by the absence of neuronal ganglion cells in a distal portion of the intestinal tract [1]. In 1691, Frederick Ruysch described the disease as congenital megacolon. HSCR-associated congenital anomalies have been reported in 5–32% of affected patients [2]. The clinical symptoms of HSCR are usually evident in the neonatal period. However, in some cases where the extent of the aganglionic segment is short, symptoms may become clinically relevant later in childhood [3]. HSCR is one of the most difficult diseases to identify in pediatric surgery due to its multiple clinical, histological and radiological variations [2, 3]. The goal of surgical management is to remove the aganglionic segment and reconstruct the intestinal tract through techniques such as Swenson, Duhamel and Soave [4]. The following case consists of a 4-year-old patient with a chronic presentation of constipation secondary to ultrashort-segment Hirschsprung disease.
INTRODUCTION
Hirschsprung disease results from the congenital deficiency of the Meissner and Auerbach plexuses in the submucosa and myenteric layer, respectively [5]. In 80–85% of cases, the disease is present as short-segment disease, 20% as long-segment, 3–8% as total colonic aganglionosis and the incidence is even lower for USHD [6]. HSCR disease in neonates often manifests with delayed passage of meconium after birth, intestinal obstruction and constipation, which can progress to abdominal distention and bilious vomits. When the patient presents with USHD, occasional defecation can be possible, making diagnosis more difficult [5, 6]. Hirschsprung-associated enterocolitis (HAEC) is a serious complication of HSCR that occurs in nearly 16% of patients and contributes to 50% of mortality. HAEC usually occurs 2–4 weeks after birth, but can also present in older patients or after treatment with the pull-through procedure. Urogenital complications such as vesicoureteral reflux and hydronephrosis can develop secondary to chronic obstruction [2, 6]. Numerous tools are useful for HSCR diagnosis, yet the goal standard is rectal suction biopsy. The primary treatment is surgical resection of the aganglionic segment with anastomosis of the proximal normal colon to the rectum [5, 6].
CASE PRESENTATION
The clinical case consisted of a 4-year-old female patient who presented with long-standing constipation, which afflicted her since birth. The mother referred that the patient could only defecate 2 to 3 times per week, requiring the administration of rectal enemas, lactulose and plum juice. Upon physical examination, the patient presented increased frequency of intestinal sounds, no abdominal tenderness on palpation, tympanic sound to percussion, abdominal distention and a positive blast sign. A water-soluble contrast enema was performed to evaluate the colonic segments and rectum. The study displayed a distal narrow segment with proximal distention representing the pathognomonic transitional zone (Figs 1 and 2). Rectal biopsies were taken at 3, 5 and 10 cm from the anal margin. The biopsy at 3 cm showed hyperplasia of the muscularis mucosae and absence of the submucosal and myenteric nerve plexuses. However, the other two segments that were taken showed no abnormal findings. According to these findings, the patient was diagnosed with USHD. Treatment consisted of transanal myectomy of the internal anal sphincter.
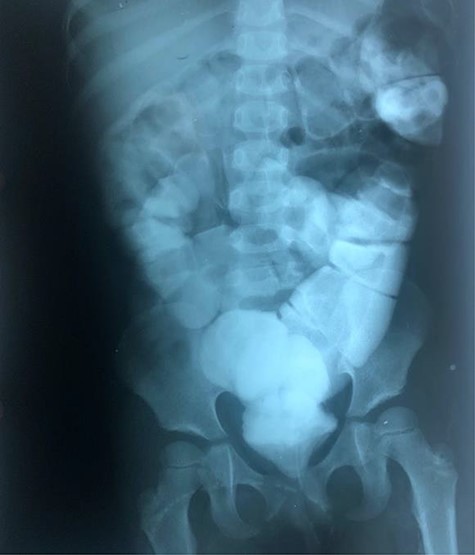
Radiographic abdominal anteroposterior projection of water-soluble contrast enema.
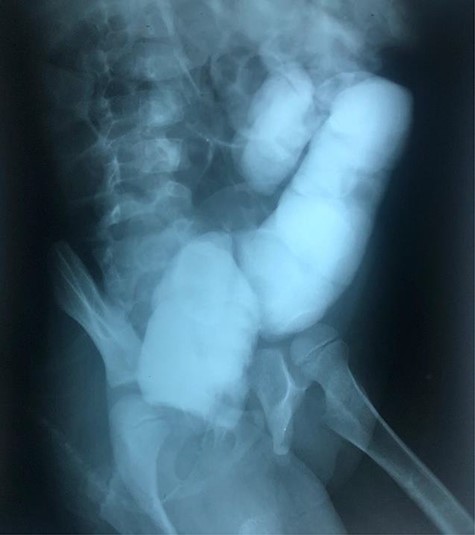
Radiographic abdominal lateral view of water-soluble contrast enema.
DISCUSSION
Hirschsprung disease is a congenital disorder of the enteric nervous system characterized by distal aganglionosis [7]. Classification is established according to the extent of the aganglionic area as short segment, long-segment, total colonic and ultra-short segment. The transition zone is the portion of the bowel proximal to the aganglionic region. Epidemiologic data suggest that 1 in 5000 live births present with HSCR with a 4:1 male predominance [6, 7]. Down syndrome, dominant sensorineural deafness, neurofibromatosis, pheochromocytoma and neuroblastoma have all been linked to syndromic HSCR. Significant susceptibility genes include RET and EDNRB [8]. This disease can generate obstruction in the neonate, which manifests as abdominal distension, bilious vomiting and feeding intolerance. Delayed meconium passage after 24 hours from birth is characteristic but can be absent in nearly 10% of patients with HSCR. Patients diagnosed later in life often have a short-segment variant of the disease, and their clinical presentation usually consists of chronic constipation and distention, vomiting and failure to thrive [4, 6, 8].
Diagnosis can be made by a contrast enema, biopsy and manometry. The first step is a water-soluble contrast enema; the pathognomonic finding is a transitional zone between normal and aganglionic segments, which is present in 70–90% of cases. Anorectal manometry may help with the diagnosis; however, it is only reliable when the recto-enteric reflex is present, after neonatal day 12 [6, 9]. The diagnosis can be confirmed with suction biopsies of the rectal mucosa and submucosa. This method is preferred because of its safety and because there is no need for general anesthesia [9]. Acetylcholinesterase staining techniques have been used to demonstrate an increase in neurofibrils in the lamina propria and muscularis mucosae. USHD is diagnosed with a documented aganglionic segment of less than 1 to 2 cm. Surgery is required after the diagnosis of HSCR has been established. The most commonly used techniques are Swenson, Duhamel and Soave [2, 4, 9]. For USHD, experts have described two different surgical approaches: anal sphincter myectomy and excision of the aganglionic segment with pull-through reconstruction [4, 9].
CONCLUSION
USHD is a rare variant that often manifests as chronic constipation, and occasional defecation makes the diagnosis difficult. Different modalities of diagnosis are available; however, the initial step is a water-soluble contrast enema, and the gold standard is a biopsy; hence, these were the two studies utilized to diagnose the patient [4, 6, 9]. According to the biopsy and contrast enema, the patient was diagnosed with USHD presenting chronic constipation and distention. The treatment is controversial, and some authors recommend a simple anal sphincter myectomy, which was performed in this patient.
CONFLICT OF INTEREST STATEMENT
None declared.
FUNDING
None.

{kind=link}
{kind=link}