





Abstract
An 8-year-old girl was admitted with four limb weakness for 2 months. Hyperactive reflexes were observed in all four limbs, and positive Hoffmann’s signs were revealed. An MRI spine with the coronal slide detected two tumors, first in the intradural and intramedullary space at the C2–C4 level and the second in the intradural and extramedullary space at the C5–C7 level. Axial T1W contrast MRI brain showed a tumor in the left parietal region. The patient underwent spine surgery first and following by brain surgery. No invasive lesions were remarked during surgery. Histological findings showed meningothelial meningioma and ependymoma from the spine and rhabdoid meningioma from the brain. Two months after the second surgery, the patient recovered fully with no symptoms and was able to participate in all regular activities in life. This work is the first report of a mixed tumor with distinct spinal meningioma and ependymoma components occurring in the cervical spinal cord at the C2–C7 level and coexisting with cranial meningioma. The remarkable result was that the patient fully recovered without any symptoms after receiving two surgeries.
INTRODUCTION
Multiple tumors of the central nervous system are associated with gene disorder, especially the mutation of NF2 gene located on chromosome 22q12 [1]. Without definite evidence of NF-2 mutation, the coexistence of two different primary tumors with different histogeneses is uncommon. Limited cases of multiple tumors without NF-2 mutation, such as synchronous intracranial and spinal meningioma, have been reported [2]. In literature, cases with numerous tumors of different pathologies at various locations are rare. Here, we presented a case of mixed tumor with distinct spinal meningioma (meningothelial meningioma) and ependymoma components occurring in the cervical spinal cord at the C2–C7 level and coexisting with intracranial meningioma (rhabdoid meningioma). To our best knowledge, this type of case has never been reported.
CASE REPORT
An 8-year-old girl was admitted with four limb weakness for 2 months. These symptoms were gradual at onset but had progressively increased in severity. No related family and epidemiological histories were noted, and development was normal. Neurological examination on admission in July 2018 revealed grade 4/5 on the left side and grade 3/5 on the right side. Hyperactive reflexes were observed in all four limbs, and positive Hoffmann’s signs were revealed. However, no sensory deficit, urinary or fecal incontinence, visual impairment, focal neurological symptoms and muscle atrophy were noted.
An MRI spine (Fig. 1) with the coronal slide detected two tumors, first in the intradural and intramedullary space at the C2–C4 level and the second in the intradural and extramedullary space at the C5–C7 level. Sagittal T1W contrast MRI spine revealed another enhanced broad-based intradural/extramedullary space at the C6–C7 level. Axial T1W contrast MRI brain (Fig. 2) showed a tumor in the left parietal region.
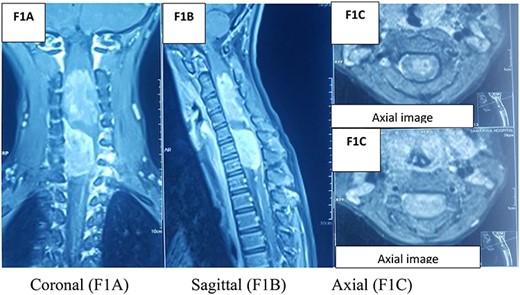
MRI spine.
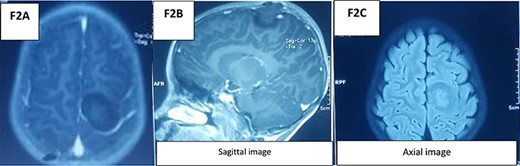
MRI brain.
The management was a step-by-step surgery.
(i) The first surgery was performed to remove the tumors in the spine. The extramedullary–intradural tumor at C5–C7 was removed via laminectomy at C2–C7, and histopathological examination confirmed it as meningothelial meningioma (WHO Grade I). The intramedullary–intradural tumor at C2–C4 was also removed, and histopathological examination classified it as ependymoma. With C2–C7 laminectomy, fusion was performed intraoperatively with pedicle screws to ensure stability and avoid spinal deformity.
After the spine surgery, the symptoms did not immediately improve, and the patient eventually presented with muscle strength grade 2/5, hyperalgesia and urinary and fecal incontinence. However, the only sign that remained 20 days after surgery was muscle strength grade 4/5. Temporary neurological deterioration occurred following the resection of the spinal tumors. Methylprednisolone with a dosage of 40 mg per day was used for 10 consecutive days. The spinal hematoma was not a substantial concern because the intraoperative bleeding was controlled, the drainage fluid was minimal and neurological improvement was observed day after day.
(ii) Twenty days after the first surgery, the patient underwent brain surgery via craniotomy. Histopathological study revealed rhabdoid meningioma (WHO Grade II). Three days after brain surgery, the surgeon discovered brain swelling that must be decompressed by the second surgery. The patient had cranioplasty 1 year after the decompression surgery and appeared healthy in clinical and radiological aspects.
No invasive lesions were remarked during surgery. The patient was given antibiotics, analgesics and anticonvulsants during the postoperative period of the two operations. No adverse event occurred, and good recovery was observed. At discharge, the patient was in good condition. Two months after the second surgery, the patient recovered fully with no symptoms and was able to participate in all regular activities in life.
Histological findings
After surgery, the tumor at C5–C7 showed meningothelial meningioma (Fig. 3). The second tumor in the spine at C2–C4 was ependymoma (Fig. 4). Tumor in the brain showed rhabdoid meningioma (Fig. 5).
![Histologic description of meningothelial meningioma (WHO Grade I). M1 and M2 (hematoxylin and eosin [H&E] ×50): syncytial growth of meningothelial cells with sparse psammoma bodies; uniform tumor cells that form lobules surrounded by thin collagenous septae. Within the lobules, epithelioid tumor cells have fuzzy ill-defined cell borders. M3 and M4 (H&E ×200): characteristic nuclear include clear spaces (that seem empty of karyoplasm) and rounded eosinophilic cytoplasmic protrusions.](https://oupdevcdn.silverchair-staging.com/oup/backfile/Content_public/Journal/jscr/2020/8/10.1093_jscr_rjaa267/1/m_rjaa267f3.jpeg?Expires=1788202767&Signature=XXHGcAMDHxj7CKJB4csuQu4LUMdCB7FYFFiFNtkJIDufsyNPRm85p4SYrKb8qkQwT7mrJwiipQiDiMlb1XYeGLAZnxwAJEuZqQZnqX39REYpQQyX4KoQgifWScwjJbS2Ekw~Qm1GslAnn6YVX10gzMkxDBvazzfBBNF43kgcu2m9GJCfAloR2TWsOPoez7M7SOg98d5Ij7HLY2QGv5-2pY8nIKl3~bZqvaRF6xZcwSuMh8pFzjkFNwaXUf7yjkPOsFfwGXUPRKTxBuAsaw9NlFYGPKD01qcqaOGs-0mOjB-RKKmjIIyFHJ6YmUUMjRkRFz0MjYqRmvMy98sb5o44KQ__&Key-Pair-Id=APKAIYYTVHKX7JZB5EAA)
Histologic description of meningothelial meningioma (WHO Grade I). M1 and M2 (hematoxylin and eosin [H&E] ×50): syncytial growth of meningothelial cells with sparse psammoma bodies; uniform tumor cells that form lobules surrounded by thin collagenous septae. Within the lobules, epithelioid tumor cells have fuzzy ill-defined cell borders. M3 and M4 (H&E ×200): characteristic nuclear include clear spaces (that seem empty of karyoplasm) and rounded eosinophilic cytoplasmic protrusions.
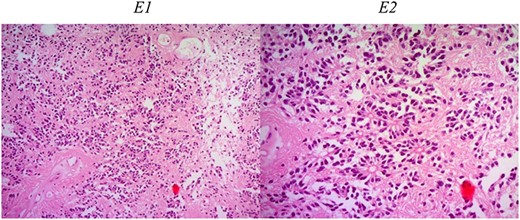
Histologic description of ependymoma component. E1 (H&E ×50): moderately cellular with round/oval nuclei having discrete interface with surrounding parenchyma. E2 (H&E ×200): monomorphic cells with salt and pepper chromatin. Key histological features are perivascular pseudorosettes and ependymal rosettes.
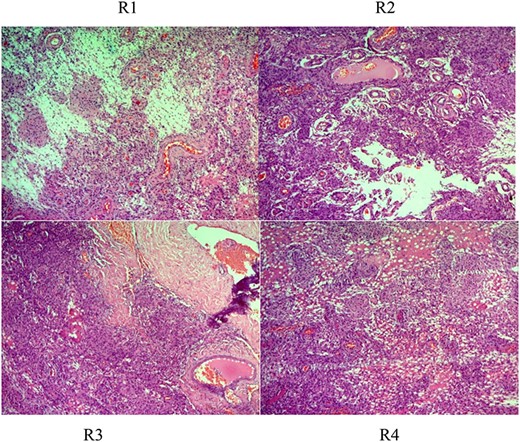
Histologic description of rhabdoid meningioma (brain) (WHO Grade II). R1–4 (H&E ×50, ×100, ×200): sheets of loosely cohesive cells with clear nuclei and pleomorphism, retaining meningothelial features (whorl formation).
DISCUSSION
Multiple tumors in the central nervous system are differently termed as coexisting tumors, collision tumors and concurrent tumors. This condition usually occurs with gene mutations, such as that of NF2 gene that is related to multiple meningiomas. NF2 gene mutation is characterized by hamartomatous and/or neoplastic proliferation of Schwann, meningothelial and glial cells [3]. Our patient had developed a meningothelial meningioma that is unfit with NF2 diseases. In addition, two different meningiomas in two varying locations (cranial and spinal) were recorded. Smith et al. showed that SMARCE1 loss is relevant to cranial and spinal meningiomas [4], but this gene mutation is still inadequately evaluated. In particular, meningothelial meningioma WHO Grade I is associated with the mutation of AKT1 (13%), NF2 (22%) and SMO (16%) [5]. Moreover, rhabdoid meningioma located near falx cerebri shows an association with SMARCB1 [6]. In our case, the gene relation between the two types of meningioma was unclear. Moreover, ependymoma is associated with L1CAM expression and the genes RELA and C11orf 95 originating from chromosome arm 11q [7]. This finding supported the hypothesis that the three tumors are coincidental rather than gene mutations that occur through frequently mutated genes in each type of tumor. However, all of these statements are speculations. The difference in the mutated genes does not explain their hypothesis. In addition, the genetic data for the each tumor of our patient were not investigated because in Vietnam, no kits for these mutation genes are available. Our patient also showed no gene-related history. Hence, this report concentrated on the surgical perspective rather than on the gene mutation.
First-line treatments for meningioma are observation and surgery. Adjuvant therapy, such as radiotherapy/radiosurgery, is discussed for atypical and indicated for anaplastic meningioma [8]. On the basis of the histological findings (meningothelial meningioma WHO Grade I and rhabdoid meningioma WHO Grade II), surgery without adjuvant therapy was recommended. In children with Grade II spinal ependymoma, maximally safe resection is the cornerstone [9]. Depending on the residual disease and tumor grade, the precise adjuvant therapy of ependymoma is radiation therapy [10]. Histological findings of ependymoma in our case showed no malignancy. Follow-up was continued without adjuvant treatment. Two months after the second surgery and up to 24 months, the patient recovered fully with no symptoms and was able to participate in all regular activities in life.
CONCLUSION
This work is the first report of a mixed tumor with distinct spinal meningioma and ependymoma components occurring in the cervical spinal cord at the C2–C7 level and coexisting with cranial meningioma. The remarkable result was that the patient fully recovered without any symptoms after receiving two surgeries.
Conflict of Interest statement
The authors have no conflict of interest.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial or not-for-profit sectors.
Ethical approval
Our case report was not made with experimentation; we just described our clinical practice.
Consent
Written informed consent was obtained from the patient for publication of this case report and accompanying images.
Guarantor
All authors have read and approved the manuscript and accept full responsibility for the work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}