





Abstract
Paraganglioma in the spine remains a rare occurrence that is mostly benign and commonly associated with other inherited symptoms. Presentation in the pediatric population is rare with a high risk of recurrence. This case reports an unusual presentation of a slowly progressing nonfunctional thoracic paraganglioma in a 6-year-old female child that presented with mass-related symptoms sparing the spinal canal. Tumor recurred after initial video-assisted thoracoscopic surgery excision with significant involvement of the thoracic spinal canal. Patient underwent a second surgery utilizing a posterior approach and laminectomies. Succinate dehydrogenase-B gene association was confirmed through molecular testing afterward. Such tumors can be malignant with 7% present with distant metastasis. Image-based differentiation of malignant tumors remains difficult, adding to the urgency in diagnosing these tumors. Furthermore, the unlikely age presentation compounds to the challenges of the diagnostic process.x The patient remains tumor free 12 months postoperatively.
INTRODUCTION
Paraganglioma is an extra-adrenal pheochromocytoma that originates from chromaffin cells with the ability of secreting neuropeptides and catecholamines [1]. It is associated in 10–50% of cases with inherited syndromes such as Von Hippel–Lindau disease [2]. Furthermore, it occurs in the thoracic and abdominal sympathetic nerves with 15–24% present as functional paraganglioma [3]. The manifestations include symptoms related to location-dependent mass effect such as chest pain, dysphagia and hoarseness and/or sympathetic hyperactivity such as hypertension, flushing and diaphoresis [4]. Moreover, nonfunctional paragangliomas are usually found incidentally. Initial evaluation and early detection of the tumor are crucial for better long-term outcomes with complete surgical resection continuing to be the treatment of choice. Paragangliomas of the spine, although rare, are usually noted in the lumbar region. Thoracic spine involvement is unusual. The age presentation of this condition is usually reported in the adult population rather than the pediatric one [5]. This report presents a rare case of thoracic spinal paraganglioma in a pediatric patient.
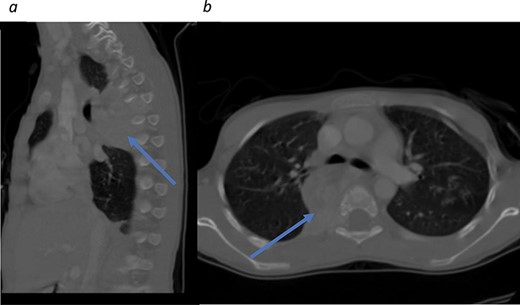
Initial CT scans. (a) Sagittal view, tumor noted in the mediastinal area between T4–T7. (b) Axial view showing tumor causing no narrowing of the spinal canal.
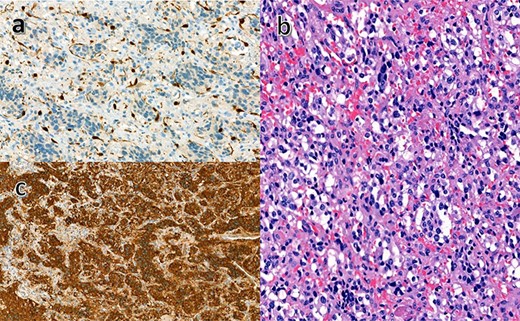
Histopathology of the excised specimen. (a) The sustentacular cells of the spinal metastases of pheochromocytoma showing characteristic staining of S100. (b) Nests of tumor cells showing significant nuclear pleomorphism with prominent nucleoli (hematoxylin and eosin). (c) Positive chromaffin cells. Chromogranin A is noted in the secretory granules (chromogranin A immunostaining).
CASE PRESENTATION
A 6-year-old female with history of on and off cough associated with chest pain was seen in another hospital and diagnosed with mediastinal mass discovered on chest X-ray. She was referred to our hospital for further investigation and management. The patient denied history of palpitation, flushing, headache or any neurological symptoms. On clinical examination, she was a stable patient with no sensory or motor deficit. Routine laboratory work was carried out and results were unremarkable. Contrast-enhanced computerized tomography (CT) scan was done to further investigate the mass and showed a highly vascular right posterior mediastinal tumor measuring 3.29 × 1.2 × 4.6 cm extending from the level of T4 down to T7 encroaching the carina and right main bronchus and sparing the vertebrae (Fig. 1). The treating team (Thoracic Surgery) and the family decided to proceed with surgery and the patient was taken for video-assisted thoracoscopic surgery (VATS) for tumor excision through lateral approach. Intraoperatively, the tumor bled profusely, and the procedure was converted to open thoracotomy to control the bleeding. The tumor was resected completely, and a specimen was sent for histopathology. Pathology report returned back to show benign paraganglioma (Fig. 2). The need for further investigations was explained, as the disease could be associated with other benign tumors that should be identified.
Appropriate tests were done (vanillylmandelic acid and norepinephrine metabolites were found to be slightly high, urine metanephrines were within normal limits, insulin-like growth factor-1 (IGF-1), IGF-binding protein, adrenocorticotropic hormone, cortisol and growth hormone levels were all normal. During her clinic follow-up 6 months post resection, a positron emission tomography (PET) scan showed hypermetabolic activity in the soft tissue at the level of T5 and adjacent right costovertebral junction with possible residual tumor and T5 involvement (Fig. 3). Magnetic resonance imaging was carried out after and showed significant narrowing of the spinal canal at the level of T5 and T6 and focal signal intensity at T5 suggestive of bone involvement (Fig. 4).
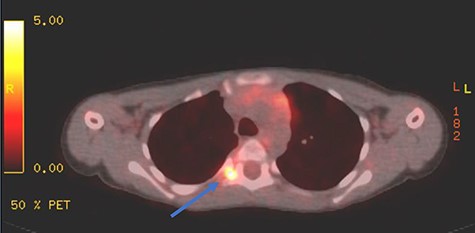
PET scan showing area of focal enhancement at the level of the tumor.
A diagnosis of recurrent paraspinal paraganglioma was made and she was admitted for tumor resection. Through posterior approach, laminectomy of T5, lower part of T4 and upper part of T6 was done (Fig. 5). The tumor was exposed and resected along with the parietal pleura. Tumor specimen was sent for histopathology, which confirmed the recurrence of the tumor.
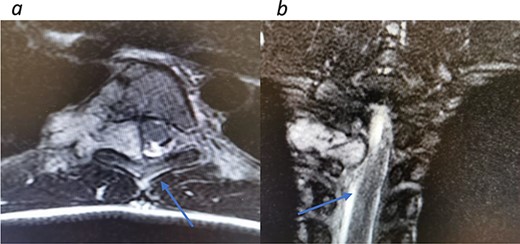
Redemonstrations of tumor post VATS. (a) Axial view tumor noted with arrow causing significant narrowing of the canal. (b) Coronal view of thoracic spine showing involvement of the spinal canal.
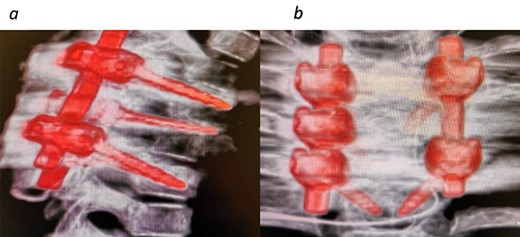
CT images 6 months post tumor removal and instrumentation. (a) Sagittal view and (b) coronal view.
The patient was referred for genetic testing to rule out underlying hereditary genetic predisposition to malignancy. The diagnosis of hereditary paraganglioma–pheochromocytoma resulted from mitochondrial succinate dehydrogenase (SDH)-B gene mutation and confirmed by molecular tests. Patient continues to have no tumor recurrence 12 months postoperatively.
DISCUSSION
Paragangliomas of the spine continue to be reported in the literature as a rare occurrence. Classified as a Grade I tumor by the World Health Organization, they are usually benign and slow-growing tumors [6]. Incidence rates of the tumor are unclear as of yet, and no more than case series reports were found in the literature. The largest of which is a recent study by Yin et al. [4] where they reviewed 18 patients over an 11-year period. Most cases present in the lumbar spine region with thoracic spine involvement rarely reported. Furthermore, most reviewed cases report a mean age of 40–50 years with pediatric involvement seldom reported. The challenge in this case is compounded by the unlikelihood of the diagnosis in a child. The authors were only able to find three reports that describe paragangliomas in the thoracic spine in children [7–9]. Such tumors, although usually benign, can be malignant with ~7% of malignant paragangliomas present with distant metastasis, which highlights the need for prompt diagnosis and complete excision of the tumor [4].
Paragangliomas with spine involvement are known to be associated familial syndromes, such as multiple endocrine neoplasia Type 2, neurofibromatosis Type 1 and Von Hippel–Lindau disease. Furthermore, germ line mutations in the mitochondrial SDH subunits (B, C and D) increase the risk for developing paragangliomas. The most commonly found mutation is SDH-B accounting for 1.7–6.7% of total cases. Moreover, patients with the mutation are usually suffering from multifocal disease, which highlights the importance of proper imaging studies [4]. Continued follow-up for these patients shows a recurrence rate of 2.2% in the literature. Multiple studies reported a recurrence after complete resection in their case series in at least one patient, whereas others reported none [4, 5, 9]. Subtotal resection, however, can increase the figure substantially. Gelabert-Gonzalez and colleagues reported a recurrence rate between 5.4 and 10.5% in their review literature of 174 cases [4, 10].
The patient presented in this report highlights many of the challenges previously described with proper and fast diagnosis of the tumor continuing to be difficult. Tumor recurrence necessitated a second surgery in this case. Furthermore, the benign nature of the patient’s tumor allowed for ample time for proper medical attention to be given, with only mass-related effects raising suspicion. Proper imaging and molecular testing can help direct further treatment to rule out malignant tendencies and multifocal disease.
CONFLICT OF INTEREST STATEMENT
None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}