





Abstract
Malignant triton tumor (MTT) is a rare subtype of malignant peripheral nerve sheath tumors (MPNSTs) histologically defined by rhabdomyoblastic differentiation. MTTs are primarily found in the head, neck, extremities and trunk, but rare cases of MTT within the buttock, the mediastinum and the retroperitoneum have also been documented. We present the case of a 47-year-old male patient who initially presented with right flank pain and hematuria in July 2019, who was found to have a large pelvic mass below peritoneal reflection. Complete resection of the mass was performed, and pathology identified the mass as a MTT.
INTRODUCTION
The incidence of malignant peripheral nerve sheath tumors (MPNSTs) in the general population is 0.001 %, accounting for only 5% of malignant soft tissue tumors [1, 2]. Many MPNSTs are associated with neurofibromatosis 1 (NF-1); the remaining cases are sporadic [2–5]. MPNSTs are derived from Schwann cells or pluripotent cells of the neural crest and are characterized by their spindle-shaped cells. In 15% of MPNSTs, other cellular components can also be observed [4, 6].
A malignant triton tumor (MTT) is a rare subtype of MPNST, histologically defined by rhabdomyoblastic differentiation. MTTs primarily arise from a peripheral nerve in the head, neck, extremities or trunk, but rare cases within the brain, buttock, viscera, mediastinum and retroperitoneum have also been documented [1, 4, 7]. MTT is aggressive and often difficult to completely resect, with poor prognosis and a reported 5-year survival of 11% [4]. Treatment is similar to that of rhabdomyosarcomas including primarily surgery with additional chemotherapy or radiation, though it is unclear whether radiation or chemotherapy improves outcomes [1, 4, 7].
CASE REPORT
A healthy 47-year-old male with past medical history significant only for hypertension and nephrolithiasis presented to Englewood Health Medical Center in October 2019 with right flank pain radiating to the pubis, hematuria, hard stool with straining and fifteen pound weight loss over 6 months. On physical examination, multiple café au lait spots, small neurofibromas on his trunk and arms and axillary and inguinal freckling were observed. On CT scan, he was found to have a 10.1-cm soft tissue mass in the right hemipelvis and a nephroureteral stent with a 3-mm stone in the proximal right ureter (Fig. 1). Further history revealed the patient had a known pelvic mass discovered on CT approximately 10 years prior (Fig. 2), which was again observed on CT in June 2019 during a hospitalization for nephrolithiasis at Hackensack University Medical Center (Fig. 3); he had been lost to follow-up.
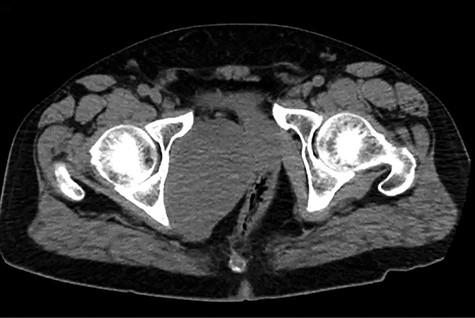
EHMC 10/21/19: 10.0 × 6.5 × 7.5-cm-sized right pelvic wall soft tissue mass.
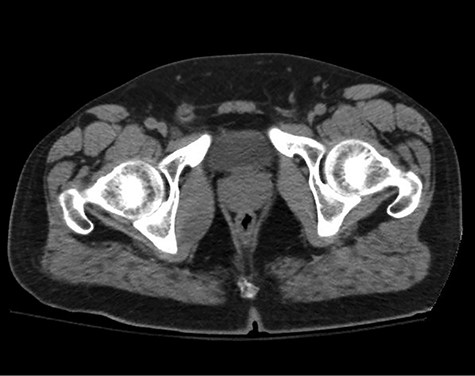
HUMC CT 11/14/09: 3.3 × 6.6 × 3.0-cm-sized right pelvic wall soft tissue mass.
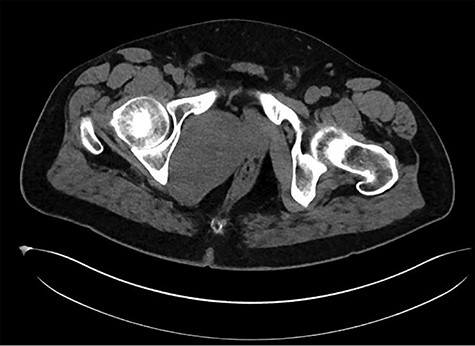
HUMC 6/1/19: 9.7 × 6.3 × 7.5-cm-sized right pelvic wall soft tissue mass.
During this hospitalization, colonoscopy with biopsy of the mass and partial polypectomy of a large benign-appearing polyp in the ascending colon was performed. Pathology revealed spindle cells and a tubular adenoma with intramucosal adenocarcinoma, respectively. He was discharged home with scheduled follow-up for preoperative planning for robotic resection of the pelvic mass via abdominoperineal approach. However, days later, he returned to the emergency department with worsening right flank pain. He was taken to the operating room for mass resection. Rectal examination revealed a palpable large pelvic mass displacing but not invading the rectum. Initial laparoscopy revealed no abnormality in the small or large intestine, omentum, liver or spleen. Robotic approach was utilized to enter the mesorectal space posterior to the rectum, taking care to preserve the ureters and hypogastric nerves (Fig. 4). Robotic dissection continued to open the anterior peritoneal reflection, where a large tumor was noted abutting the right side of the rectum, pelvic bone and obturator canal. The mass was carefully separated from these structures using sharp dissection and electrocautery (Fig. 5). The urology team then continued dissection, freeing the mass from the prostate, bladder and right seminal vesicle. The tumor was removed through a Pfannenstiel incision covered by a GelPort. The incisions were closed. The patient was extubated in the OR, having tolerated the procedure well. His postoperative course was only remarkable for a brief period of urinary retention, which resolved spontaneously. He was discharged home on postoperative day 3 with scheduled follow-up appointments with urology and colorectal surgery.
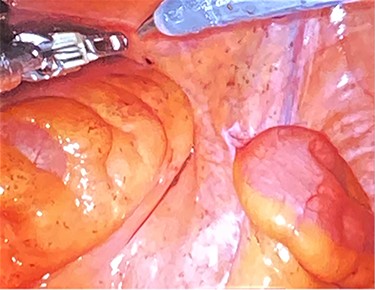
Dissection into the mesorectal space to open the anterior peritoneal reflection.
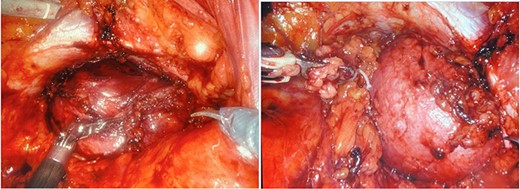
a and b. Robotic dissection of the mass from the right side of the rectum, pelvic bone, obturator canal, prostate, bladder and right seminal vesicle.
The pathology revealed a 282-g nodular tumor measuring 10.7 × 8.5 × 7 cm that was grossly tan, fibrotic and firm with focal yellow discoloration and soft flesh areas (Fig. 6). Microscopic analysis revealed high-grade spindle cell sarcoma arranged in intersecting fascicles with extensive areas of necrosis (Fig. 7). The lesional cells had fibrillary eosinophilic cytoplasm and evoid nuclei with nuclear pleomorphism (Fig. 8). Areas of rhabdomyoblastic and chondroid differentiation were also noted (Figs 9 and 10). Immunostaining was positive for desmin, focally for CD 34 and Bcl2, while Cam 5.2, CD117, S-100, SMA and HME45 immunostains show patchy staining for myogenin. Expression of H3K27me3 was completely lost in the tumor cells. MyoD1, SOX10, MDM2 and CDK4 were negative. It was identified as a high-grade MPNST with heterologous rhabdomyosarcomatous differentiation, i.e. MTT. All margins were negative.
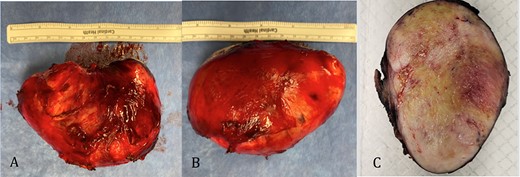
Gross pathology: A and B. 282-g nodular tumor mass, measuring 10.7 × 8.5 × 7 cm. The external surface covered by tan, fibrous tissue. Attached irregular, fibrous tissue measuring 7 × 2.5 cm. C. Black ink was applied on the external surface. Cut surfaces are light tan, firm, fibrotic appearing with focal yellow discoloration and soft flesh areas.
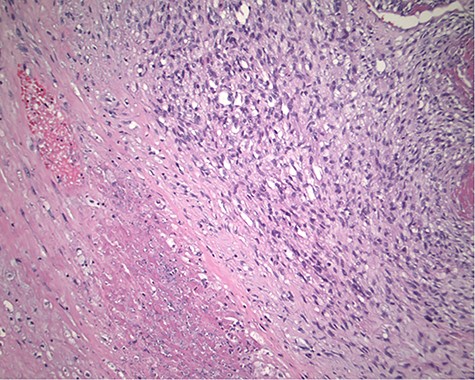
Magnification 100×. Cellular spindle cell neoplasm with focal necrosis.
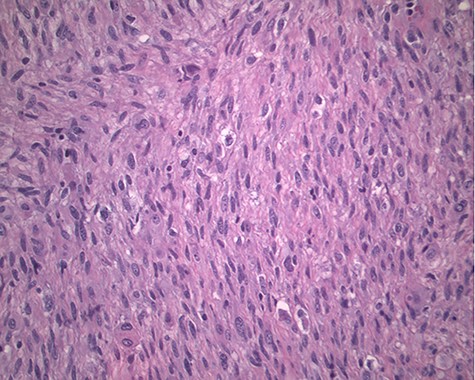
Magnification 200×. Fascicles of spindle cells with ovoid nuclei and eosinophilic nuclei.
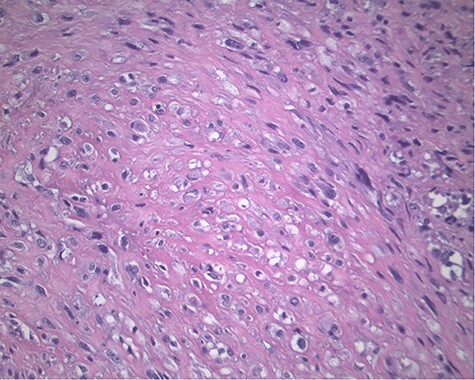
Magnification 200× Spindle cells with mild to moderate nuclear pleomorphism and focal rhabdomyoblastic differentiation.
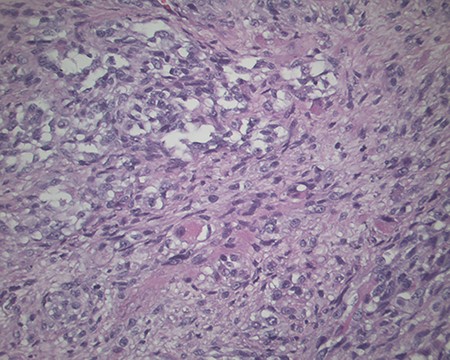
Magnification 200×. Chondroid differentiation.
DISCUSSION
MPNSTs with rhabdomyosarcomatous elements were first described by Masson in 1932 [8]. In 1973, Woodruff et al. named and proposed criteria to classify a mass as a MTT, including tumors in patients with or without NF-1 that are microscopically compatible with a malignant schwannoma and contain rhabdomyoblasts [9].
While MTT has been documented in many age groups, the mean age of those affected is approximately 32 years with equal sex distribution. 44–69% of cases are in patients with NF-1, and those with NF-1 are more likely to present at a younger age [4].
MTT is a highly aggressive tumor with low overall survival rates, high rates of distant metastasis and local recurrence [4]. The size, grade and location of the tumor in addition to extent of the excision appear to be primary factors affecting survival, though most patients do not survive beyond a year of diagnosis despite treatment [2].
The presence of immunohistochemistry reactivity to S-100, SOX10, Leu-7 or GFAP indicates nerve sheath differentiation. However, only a subset of MPNSTs express these Schwann cell markers, and therefore, histologic features in combination with immunoreactivity are important in the diagnosis of MPNSTs [4–6]. Rhabdomyoblastic components are identified with positive immunoreactivity to desmin, muscle-specific actin, myosin, vimentin and myoglobulin. In addition, loss of H3K27me3 has been shown to be specific for MPNST [10].
The present case was a 47-year-old male, who was diagnosed with NF-1 by our team upon presentation to EHMC. He presented with a large mass below the peritoneal reflection with involvement of adjacent organs. The pathology from our specimen was immunoreactive to S-100 and desmin, and expression of H3K27me3 expression was completely lost, indicating a MTT. As is typical of patients with retroperitoneal MTTs, the insidious onset resulted in late presentation. The large size of the mass and involvement of surrounding organs contribute to a poor prognosis despite complete resection of the mass and negative pathologic margins. Treatment modalities are not well established due to the rarity of MTT, contributing to the poor prognosis.
Dr. Seddighzadeh is a resident physician who performed the literature review and wrote the manuscript as the primary author. Credited with investigation, writing-original draft, project administration, visualization.
Dr. Brower is an oncologic surgeon and the Chief of Surgical Oncology and HPB Surgery at Englewood Health, who provided counseling and input related to the oncology aspects of the submission. Credited with writing-review and editing, resources, supervision.
Dr. Tzeng is the pathologist involved in the case who provided gross and histologic analysis and photos for the submission. Credited with resources, writing-review and editing.
Dr. Serur is a colorectal surgeon and the Chief of Colon and Rectal Surgery at Englewood Health. She is the primary author’s advisor who performed the resection and provided input and expertise for the submission from a colorectal surgery standpoint. Credited with conceptualization, writing-review and editing, resources, supervision.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}