





Abstract
Extraskeletal myxoid chondrosarcoma is a rare form of malignant mesenchymal neoplasm mainly localized into the limbs, particularly in the thigh and popliteal fossa. It has been classified as a low-grade sarcoma so far, but it shows a tendency to relapse and metastasize. In the early stage of disease, surgery represents the only chance of cure. In case of diffuse metastatic disease, systemic chemotherapy with anthracyclines is the standard of care. In this paper, we present a case of a patient affected by this rare disease and the analysis of radiological, surgical and histopathological aspects.
INTRODUCTION
Extraskeletal myxoid chondrosarcoma (EMC) is a form of sarcoma mainly localized into the limbs [1]. Firstly described by Stout et al., it has been classified as low-grade soft tissue sarcoma with uncertain differentiation by the World Health Organization Classification of Tumors of Soft Tissue and Bone. It is distinguished from other sarcomas by its unique histology and characteristic chromosomal translocation, typically t (9, 22)(q22; q12.2) and the EWSR1/NR4A3 fusion [1–4]. This tumor is rare and its prevalence is around 1/1000 000 people. Myxoid chondrosarcoma shows an indolent behavior but, after a long-term follow-up, local and distant progressions of disease are demonstrated in more than 40% of patients with a 10 years survival of 65–88% [4–6]. The most common site of distant metastases is the lung, but soft tissues and lymph nodes also can be involved [1, 5, 7]. To treat distant metastatic disease or local recurrence, surgery, radiotherapy and/or systemic chemotherapy have been proposed [6–9]. For patients who have chondrosarcoma in the unresectable setting, antiangiogenic agents are reportedly effective.
CASE PRESENTATION
The patient, a 55-year-old woman, has been complaining of vague abdominal pain and severe constipation lasting for 3 months. She was admitted to the gynecological department, where an abdominal ultrasound scan was made and showed a large mass of 10 centimeters in the retroperitoneal space. After this evaluation, computed tomography (CT) scan and a magnetic resonance (MR) were performed. Laboratory tests were not suggestive for tumoral disease, being neoplastic markers CEA, CA 19.9, CA 15.3 and CA125 normal in value.
Concerning imaging description, the preliminary 64-raw MD-CT (Fig. 1) found a left-sided solid hypoattenuating, well-defined pelvic mass, with poor and inhomogeneous contrast enhancement. A similar mass coexisted in the intermuscular space between left gluteus medius and gluteus minimus.
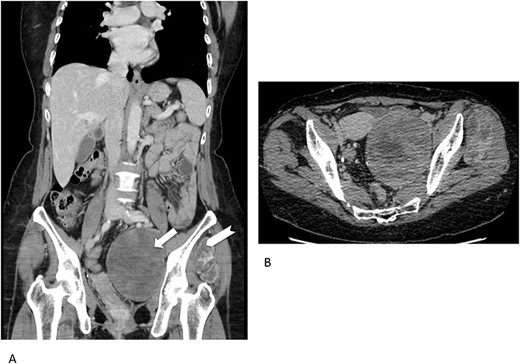
Multidetector computed tomography appearance of the lesions. Coronal multiplanar reformation (A) of the portal venous phase scan shows two different left-sided solid hypoattenuating masses located in the pelvis (arrow) and intermuscular space between the gluteus medius muscle and gluteus minimus muscle (arrowhaed). Progressive, slow and inhomogeneous contrast-enhancement occurred (B), as better appreciated in the transverse image acquired in the delayed phase.
On 1.5T MR (Fig. 2), masses were slight hypointense as compared to muscle on T1-weighted imaging, with sparse hyperintense foci suggesting the presence of hemorrhage, especially in the pelvic one. Signal intensity was predominantly slightly higher than fat on T2-weighted images, with internal low-signal septa, showing restricted diffusion and minimal contrast-enhancement. The hyperintense component was characterized by inhomogeneous contrast enhancement. Of note, there was some asymmetry between the lesions, with the pelvic one showing more inhomogeneity on T2-weighted imaging and less intense contrast-enhancement as compared to the extrapelvic one. Overall, MR findings were in line with previously reported findings [5]. At radiological scans, no distant metastases were documented in the liver or lungs.
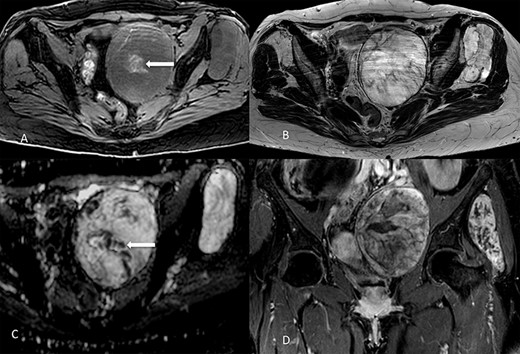
Lesions appearance on MR imaging. On transverse fat-saturated volumetric gradient echo T1-weighted imaging (A) both masses appeared slightly hypointense compared to muscle, with hyperintense areas in the larger one (arrow) suggesting the presence of haemorrhagic foci. Lesions were hyperintense on transverse turbo spin-echo T2-weighted imaging (B), in line with the presence of myxoid content, showing inhomogeneous appearance due to low-signal septa showing restricted diffusion on the apparent diffusion coefficient map (arrow in C). Contrast enhancement was inhomogeneous, as shown in the coronal fat-saturated post-contrast image (D). Masses showed well-defined margins, with no invasion of the adjacent structures.
No preoperative bioptical samples were performed and, following a multidisciplinary oncological meeting, the patient was planned for surgical procedure.
Being this sarcoma localized in two different sites, pelvic-retroperitoneal and gluteal, the operation was carried on in two surgical steps. After median laparotomy, in the left pelvic region, a mass of about 10 centimeters of maximum diameter, dislocating the bladder anteriorly, the left iliac vessels cranially and medially the sigma, was documented. Dissection and isolation of the mass from the surrounding structures were completed. A specimen of the mass capsule and myxoid material resulted to be a malignant mesenchymal neoplasia with a myxoid appearance. The mass was removed with radical intent. After its complete clearance, the entire surgical field was treated with argon-laser electrofulguration and, immediately after, with a peritoneal extensive lavage (according to Kuramoto technique) [10]. At the end of this surgical procedure, the operation was considered R0. Therefore, the patient was placed on the right flank position. An incision of the left buttock region was made, and, Deeping within the muscle planes, a lobulated mass of about 8 centimeters in diameter was found. After its complete removal, an argon-laser electrofulguration of the entire surgical area was applied. Two different surgical specimens were analyzed by the histopathological laboratory, labeled retroperitoneal-pelvic and left buttock (gluteal) tumors. At the gross examination, they both presented multiple fragments of glistening and gelatinous tissues with intratumoral hemorrhage, weighting 190 and 144 grams, respectively. Their histological features were comparable and characterized by multiple lobules with abundant myxoid-chondromyxoid matrix in which monomorphic cells with uniform nuclei and eosinophilic cytoplasm connected one another to form cords, small clusters, and complex trabecular or cribriform structures. No mitotic activity was seen; diffuse areas of hemorrhage and necrosis were identified (Fig. 3).
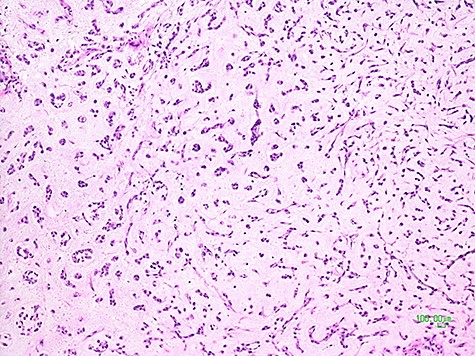
Histological picture of the tumor showing clusters and chords of medium-sized epithelioid cells immersed in a myxoid matrix (H&E, ×100).
An extensive panel of immunoistochemical markers was tested in order to better characterize the tumors. The neoplastic cells stained strongly only for vimentin; they were negative for epithelial markers (CK AE1/AE3, CK 5/6, CK7, CK19, CK20, EMA, CAM 5.2, polyclonal CEA), for muscular markers (smooth muscle actin, desmin, myogenin) and for S-100 protein, SOX10, CD34, MDM2 and Brachyury.
Considering the morphological features and the immunoistochemical profile, adenocarcinoma, myoepithelial tumors, myxoma, myxoid liposarcoma, myxofibrosarcoma and chordoma were not possible and the final diagnosis of an EMC was established.
DISCUSSION AND CONCLUSIONS
This case report is interesting for the rare double site presentation and for the pelvic localization of myxoid chondrosarcoma. The surgical workup consisted of two-step complex operations, in the pelvic area for first and then in the gluteal region. In both cases, after wide excision (Enneking classification), the tumor removal was complete. A double physical intraoperative adjuvant treatment, argon-laser electrofulguration of the entire surgical field and an extensive peritoneal lavage of the abdominal cavity, was performed. The postoperative course was uneventful. This case is the base for a multi-institutional phase II study, which intends to assess the role of adjuvant intraoperative treatment of sarcoma.
CONFLICT OF INTEREST STATEMENT
None declared.
FUNDING
None.

{kind=link}
{kind=link}
{kind=link}