





Abstract
Pulmonary lymphangioleiomyomatosis (LAM) is a rare, well-described pathology and usually is exclusive to females of a reproductive age. We present a 45 year-old lady who presented to the surgeons 1 year after an admission with acute dyspnoea following influenza infection. Initial computed tomography imaging findings demonstrated severe, heterogenous right-sided bullous emphysematous changes, but histopathological analysis of the post-operative specimen favoured a diagnosis of LAM. This case demonstrates the importance of considering LAM as a differential diagnosis for findings of unilateral emphysema or lobar emphysema, in which alpha 1-antitrypsin deficiency has been excluded and in those without a significant smoking history.
INTRODUCTION
Pulmonary lymphangioleiomyomatosis (LAM) is a rare, well-described pathology and usually is exclusive to females of a reproductive age. It can occur in conjunction with the rare genetic condition tuberous sclerosis. It tends to have a varying spectrum but generally is a slowly progressive condition ultimately culminating in respiratory failure. Prognostically, the 5-year survival of this condition is variable with 10-year survival rates between 10 and 60% [1]. Due to the progression towards respiratory failure a proportion of patients require lung transplantation [2]. The clinical presentation of these patients can have a wide degree of variance from asymptomatic patients with incidental findings on imaging to patients presenting with pneumothorax, which is the most common type of presentation [3]. It has characteristic computer tomography (CT) findings of multiple, thin-walled cysts throughout in a predominantly bilateral distribution, although unilateral distribution has been described. Furthermore, LAM has fairly consistent phenotypical appearances intra-operatively with multiple diffuse blebs visible on the surface of the lung [4]. We describe an unusual case of a middle-aged female presenting with progressive symptoms and imaging findings that were not characteristic of pulmonary LAM and in fact mistaken for emphysema with a unilateral unusual heterogenous distribution.
CASE REPORT
We present a 45 year-old lady who had initially been seen by the respiratory physicians following an admission with acute dyspnoea following influenza infection 1-year prior to her surgery. She had undergone CT scan for abnormal chest radiograph. She had previously worked as a cleaner and was an ex-smoker of 1.5 pack years with no previous asbestos exposure or underlying genetic disease such as tuberous sclerosis or alpha-1 antitrypsin deficiency. She had been followed up in the nodule clinic (a follow-up clinic run by respiratory physicians to monitor solitary pulmonary nodules) with a provisional diagnosis of chronic obstructive pulmonary disease. She had progressive dyspnoea and occasional lower respiratory tract infections treated with courses of antibiotics. Her co-morbidities included previous alcohol dependence and depression. Pre-operative pulmonary function tests demonstrated an FEV1 of 1.53 (53% of predicted), FVC of 2.52 (79% of predicted), diffusing capacity of lungs for carbon monoxide (DLCO) of 5.25 (62% of predicted) and 6 minute walking distance of 414 m. Initial CT imaging findings demonstrated severe, heterogenous right-sided bullous emphysematous changes with a basal predominance and multiple large bullae in the lower lobe (Fig. 1) and a benign inflammatory nodule, which was 7 mm in size in the right upper zone presumably right upper lobe. There was associated hyperinflation causing mediastinal shift to the left with complete right middle lobe collapse and marked compression of the right upper and middle lobes. Due to her increasing symptoms and progressive solitary nodule (Fig. 2) in the right upper zone she was referred for a surgical opinion. Pre-operative echocardiography did not demonstrate any right ventricular dilatation or impairment and no pulmonary hypertension. She was counselled regarding the progressive symptoms, lung function and risk of pneumothoraces and scheduled for right thoracotomy and lower lobectomy. Operative findings were that of large bullous disease affecting the right lower lobe (Fig. 3) with subtotal destruction of the lung parenchyma of the lower lobe. The nodule was present in the apical segment of the lower lobe instead of the upper lobe as expected. Right lower lobectomy was completed un-eventfully and the specimen was sent for histology. The upper and middle lobes were hypoplastic and the post lobectomy space management was a challenge. Post-operatively recovery was complicated by prolonged post-operative air leak managed conservatively and once this eventually settled, the patient was discharged following removal of the chest drain. Histopathological analysis of the right lower lobe specimen demonstrated several large thin-walled cysts with interstitial clusters of spindle cells bordering the cyst and immuno-histological markers (increased expression of VEGF-R3, Podoplanin, LYVE-1 and PROX-1) confirming a diagnosis of LAM. The patient was seen in the thoracic surgery follow-up clinic and a CT abdomen/pelvis was organized to rule out renal angiomyolipomas, associated with LAM in tuberous sclerosis. Referrals were made to the respiratory physicians and the National LAM centre for ongoing management.
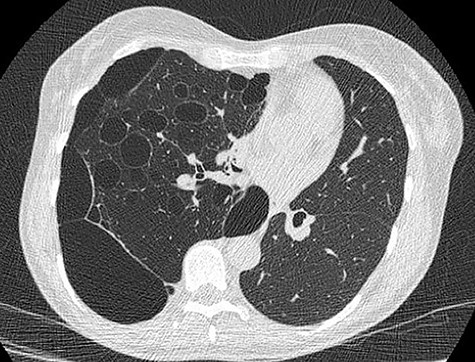
CT scan demonstrating heterogeneous bullous emphysema with hyperinflation of the right lung and mediastinal shift.
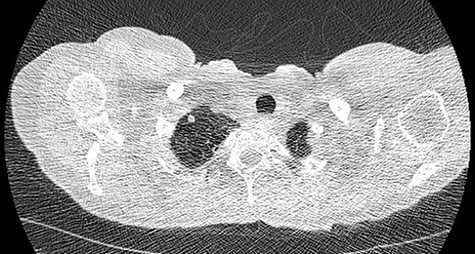
CT scan demonstrating small nodule in the right upper zone.
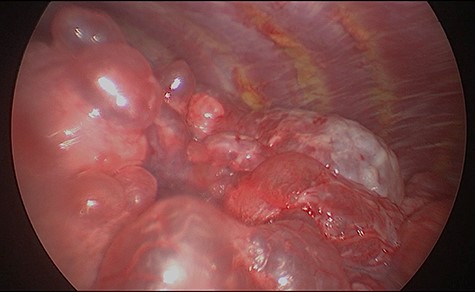
Intra-operative appearances of the right lower lobe demonstrating multiple bullae and parenchymal destruction.
DISCUSSION
LAM is a rare, well-established condition predominantly in women with variant presentation and progression [3]. It has one of the strongest gender predispositions of any extragenital human disease. The incidence in the UK and the USA is 3.4–7.8 per million [5]. This condition is often discovered incidentally or in a certain demographic of young female patients of reproductive age presenting with pneumothorax. There are sparse reports commenting on the unilateral presentation of LAM, but these were appearances described as multiple bilateral thin-walled cysts without lobar predilection or demarcation but widespread interlobular septal thickening [4]. Cyst size has been demonstrated to be a pertinent factor in LAM with respect to increased likelihood of pneumothorax, in fact a cyst size of >0.5 cm was stated to be significant [6]. Pertaining to our case, radiological CT appearances were demonstrated to be more consistent with heterogenous unilateral emphysema with a basal predominance and a nodule was reported in the upper lobe. The cysts were much larger than 0.5 cm and patient had no incidence of pneumothorax. Another confounding factor was a relatively low smoking history of 1.5 pack years with well-preserved lung function. However, the histopathological findings were confirmatory of a diagnosis of LAM. Furthermore, intra-operative findings were not classical of LAM and macroscopically the lung parenchyma was destroyed with appearances similar to that of severe bullous emphysema. Interestingly, the right upper and middle lobes were hypoplastic suggesting unknown congenital aetiology.
In conclusion, this case demonstrates the importance of considering LAM as a differential diagnosis for CT findings of unilateral emphysema or lobar emphysema in which alpha 1-antitrypsin deficiency has been excluded and in those without a significant smoking history. Furthermore, careful evaluation of all lobes intra-operatively is mandatory when considering this diagnosis in patients of reproductive age or with this type of presentation, especially if any adjunctive pleural procedures are performed concurrently as this may complicate future therapeutic options like transplantation. Early diagnosis and systemic treatments with mTOR-2 inhibitors like sirolimus or rapamycin may effectively halt or delay the debilitating disease process [7].
CONFLICT OF INTEREST STATEMENT
None declared.
FUNDING
None.

{kind=link}
{kind=link}
{kind=link}