





Abstract
Merkel cell carcinoma is a rare neuroendocrine malignancy that arises from the dermis, in cases of immunocompromised, middle-aged patients or on skin exposed to sunlight. It only rarely presents in extra-cutaneous locations. We present the case of a 63-year-old female with a mass in the adipose tissue of the upper arm, without skin involvement and concurrent axillary lymph node enlargement. She was treated with wide excision and lymph node dissection; pathology led to the diagnosis of Merkel cell carcinoma, and she was subsequently submitted to adjuvant radiotherapy. No signs of recurrence are present 8 years postoperatively. Primary Merkel cell carcinoma can rarely be located in the adipose tissue without skin involvement. In cases of high suspicion, preoperative MRI scan can show the extent of the lesion, as well as lymph metastases.
INTRODUCTION
Merkel cell carcinoma (MCC) is a rare skin malignancy of poor prognosis, arising from neuroendocrine cells of the dermis. It usually presents in patients between 60 and 80 years of age, and on skin exposed to the sun [1]; risk factors also include infection from polyomavirus, chronic inflammatory disease and immunosuppression [2]. In cases of organ-transplant recipients, patients with HIV infection and B-cell malignancy, occurrence of MCC in younger ages has been described [3].
The tumor typically presents as a rapidly growing, painless, intracutaneous mass with a color that varies between normal to bluish red. It is most commonly located on the head and neck (50.8%), whereas only 33.7% in the limbs. It is considered that only 24% presents in the upper limbs and shoulder [4]. Lymph node involvement occurs in 27% of the cases, but it can also metastasize to the liver, lungs, bone and brain [5].
We present a rare case of primary MCC with an unusual location in the adipose tissue of the upper arm without skin involvement, with metastasis to the axillary lymph nodes.
CASE REPORT
A 63-year-old female patient was referred to our Department complaining of a mass in the inner side of the lower third of the right arm, that was first palpated 4 months before had doubled in size. Her medical history was free and did not mention any other symptoms.
On clinical examination, a soft, mobile, painless mass was palpated in this area, with no visible involvement of the skin, as well as enlarged axillary lymph nodes. Due to these findings, in combination with the history of sudden enlargement, an MRI scan was performed preoperatively. A lobular, heterogeneous mass of dimensions of 3.7 × 2.9 cm in the adipose tissue in the aforementioned area was present, with an increased uptake of paramagnetic contrast. The mass did not affect the adjacent muscles or bones. Enlarged axillary lymph nodes were also present. A CT-scan and PET-scan in order to exclude distant tissue involvement were then performed.
The patient was subsequently submitted to excision of the lesion on the right arm and lymph node dissection of the right axilla. Pathology examination of the mass revealed a primary MCC with neoplastic cells displaying round nuclei with scant cytoplasm, sites of necrosis, frequent mitoses and lymph cell infiltration (Fig. 1). Immunohistochemistry showed positive stain for CK20, chromogranin, NSE, EMA, and CD67, and negative stain for CD-57 and TTF-1 (Figs 2 and 3). Two out of fourteen 14 excised axillary lymph nodes were proven to be metastatic of this tumor.
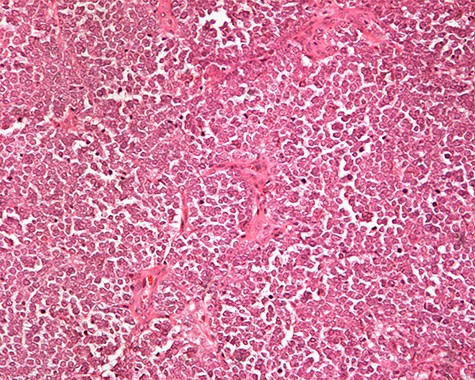
Monotonous round cells with a scant cytoplasmic rim, round vesicular nuclei, apoptotic bodies and mitoses ×20 H&E.
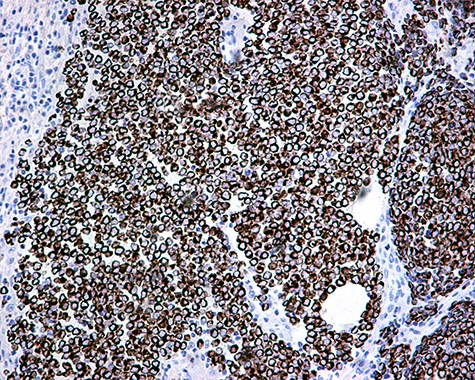
CK20 positivity: perinuclear dot-like staining ×20.
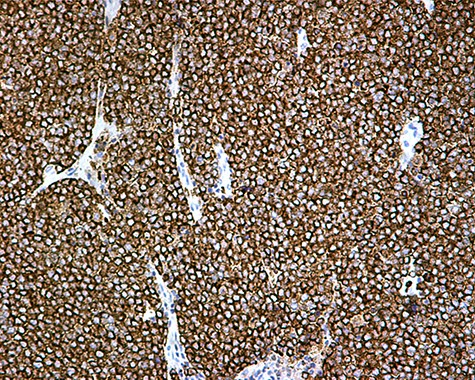
Chromogranin A positivity ×20.
The patient was further treated postoperatively with radiotherapy; follow-up examination with CT and MRI-scans after 3 months and then for 6-month intervals for the first 5 years revealed no recurrence of the disease. Eight years postoperatively, the patient remains disease free and in good health condition.
DISCUSSION
MCC is a rapidly growing, cutaneous or subcutaneous lesion, presenting as a discolored, painless nodule. It is rarely multilocular or presents with skin ulceration. Due to its clinical features, differential diagnosis includes a variety of benign and malignant conditions, such as cysts, infections and primary or metastatic neoplasia.
As these non-specific findings can be misleading, diagnosis is often delayed and cannot be based only on the clinical features. Histopathology reveals small, round-to-oval cells with a vesicular nucleus and scant cytoplasm, with multiple nucleoli, as well as sites of prominent necrosis. Factors such as tumor thickness, high mitotic rate, infiltrative growth pattern and lymphovascular invasion can be associated with poor prognosis and microscopic lymph node metastasis [6].
On immunohistochemistry, MCC is characterized by the expression of cytoskeletal keratins (CK20, CK8, CK18, CK19) and markers such as synaptophysin, BCL2, calcitonin, chromogranin A, HIP1, NSE, CD56 and somatostatin. Positivity for thyroid transcription factor 1 (TTF1), as in our case, has rarely been reported. The tumor is usually negative for vimentin, S100B and CK7 [6].
Five-year survival rate depends on the stage at diagnosis: 62.8% in stage I (<2 cm), 34.8-54.6% in stage II (>2 cm), 26.8-40.3% in stage III (nodal spread) and 13.5% in stage IV (distant metastatic spread) [7].
Due to the rarity of this malignancy, multidisciplinary management should be followed. Surgical management, with resection within clear margins and lymph node dissection, is the preferred treatment; adjuvant radiotherapy has also been proved beneficial. Combination of surgery and adjuvant radiotherapy leads to acceptable locoregional disease control in aggressive cases. However, the impact of chemotherapy on prognosis and survival has not yet been documented [8] and is reserved for palliative treatment in advanced cases.
As recurrence occurs within the first 2 years in 80% of the cases, it is advised that the patient should be clinically evaluated in 3-6=month intervals for the first 2 years and every 6-12 months for the next 3 years. CT, MRI or PET-CT scan is recommended when there is a clinical indication and should be more frequent in high-risk patients. Late risk rate is low; therefore, an annual follow-up examination is sufficient after 5 years [9].
Although a palpable mass was present in the upper limb, the surgeon should first exclude a pathology of the breast and its potential correlation to the axillary lymph nodal mass and then search for other sites of malignant tumors. It should be highlighted that the patient did not present any of the risk factors for MCC; moreover, the absence of a skin lesion could also be misleading and contribute in delayed diagnosis in a more advanced stage, and, subsequently, shorter survival. Therefore, the surgeon should be aware of ectopic MCC, as a rare cause of adipose masses, especially in cases of rapid growth. Timely imaging with simultaneous tumor excision and radical lymphadenectomy, which are also routinely advised for this tumor, are suggested to have led to initial successful treatment, which was later completed with an individualized radiotherapy [10].
In conclusion, primary ectopic or metastatic MCC in non-cutaneous sites should be taken into account in cases of rapidly growing adipose lesions, even in the absence of skin involvement, especially in high-risk patients. Combined therapy with careful follow-up leads to better prognosis.
CONFLICT OF INTEREST STATEMENT
None declared.
FUNDING
None.

{kind=link}
{kind=link}
{kind=link}