





Abstract
Multiple endocrine neoplasia type 2A (MEN2A) is a hereditary syndrome associated with tumours of the endocrine system. Principally, it is characterized by medullary thyroid carcinoma (MTC) with some individuals also developing phaeochromocytoma and hyperparathyroidism. Patients with fewer than two clinical features require either an identification of a germline rearranged in transfection mutation, or MEN2A characteristics in first-degree relatives, to establish a diagnosis of MEN2A. We present the case of a 54-year-old female with MEN2A, diagnosed by genetic screening, due to a substantial history of the disease in her ancestry. This case outlines the successful treatment of recurrent phaeochromocytoma, through the medium of radioactive iobenguane as an adjunct to surgical management. The report focuses particularly on the significance of innovative treatment strategies and forthcoming approaches to improve patient care in treating phaeochromocytoma in MEN2A.
INTRODUCTION
Multiple endocrine neoplasia type 2A (MEN2A) is a rare autosomal dominant inherited cancer syndrome (1). Most individuals with MEN2A acquire medullary thyroid carcinoma (MTC), whereas only ~50% develop phaeochromocytoma, and even fewer patients exhibit hyperparathyroidism (2).
We present the case of a 54-year-old female with MEN2A, who underwent total thyroidectomy and bilateral adrenalectomy at a young age, yet the phaeochromocytoma recurred almost 25 years later. The case illustrates the successful usage of iobenguane (MIBG) as non-surgical management in treating phaeochromocytoma. Furthermore, we analysed published literature to discuss current guidelines and updated evidence in the management of patients with MEN2A.
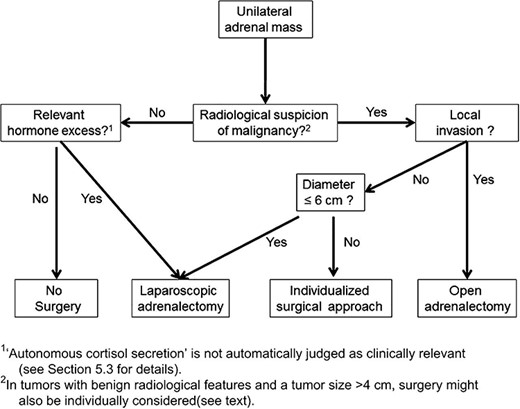
Flowchart showing surgical treatment options for adrenal masses, these same recommendations are suggested for patients with bilateral adrenal masses (4); the figure shows open adrenalectomy is only recommended if there is local invasion of the adrenal tumour, otherwise laparoscopic surgery is preferred.
CASE REPORT
A 54-year-old female patient was officially diagnosed with MEN2A at the age of 16 by a genetic screening test at an institution in London. The patient manifested a genetic mutation that placed her in a medium risk group for MTC. Suspicion of MEN2A was raised due to a substantial family history of the disease (death of five paternal relatives including the patient’s father, who died when he was 38). Genetic testing was conducted, which was a novel technique at the time and the results were not reliable enough to justify the patient enduring open surgery.
After being investigated for MTC, the patient underwent total thyroidectomy at the age of 22, despite being non-symptomatic. Following the procedure, she was started on daily oral levothyroxine supplementation. In the subsequent year, an adrenal biopsy revealed tumours which necessitated a bilateral adrenalectomy via a laparotomy. She was discharged 2 weeks later on oral fludrocortisone (once daily) and hydrocortisone (three times daily).
In 2012, the patient experienced a long period of stress and anxiety, which she believed was associated with her profession at the time. This prevailed until 2013, when she presented with these symptoms to her consultant. A blood test was requested, with the results showing elevated levels of metanephrines. This was attributable to minuscule traces of adrenal tumour that had persisted since the bilateral adrenalectomy that was performed in 1988. Beta blockers were immediately prescribed as short-term management to control metanephrine levels. The malignant tumour remnants were too small to be surgically operated on, resulting in the use of iobenguane (MIBG) as curative treatment to successfully eliminate the remaining tumour. This nuclear medicine constrained the patient to isolation for 5 days as a result of the radioactivity she was exposed to.
Currently, the patient has residual MTC due to raised levels of carcinoembryonic antigen and calcitonin but does not require treatment for this. She has also had a consistently low white blood cell count for many years but is not further susceptible to infections. Occasionally, the patient experiences episodes of tingling, light-headedness and flushing around the neck. Generally, these are provoked by tea, coffee and spicy food among other triggers. The patient is otherwise healthy and her condition is well-controlled in accordance with her ongoing treatment plan. She has follow up tests for metanephrine levels every year and a magnetic resonance imaging scan every 2 years, to monitor for recurrent phaeochromocytomas.
DISCUSSION
Clinically, MEN2A is diagnosed by the existence of two or more specific endocrine tumours (MTC, phaeochromocytoma or parathyroid hyperplasia). If less than two features are identifiable, then the presence of a germline rearranged in transfection mutation or features of MEN2A in a first-degree relative is needed (2). This case details the advantage of genetic screening to diagnose and aid in treating a patient with MEN2A, prior to developing clinical features. Since the advent of genetic testing, it is now routinely recommended to differentiate between familial and sporadic cases of specific endocrine tumours, or to diagnose children with an increased risk of MEN2A (3).
Our patient had an open surgery for her adrenalectomy in 1988, requiring hospitalization for a few weeks. Presently, laparoscopic adrenalectomies are the preferred procedure for phaeochromocytomas as they are associated with a significantly lower morbidity, shorter hospitalization and a faster recovery rate than open laparotomies (Fig. 1) (4). A new milestone in minimally invasive endocrine surgery could be attained with retroperitoneoscopic adrenalectomies. This procedure has demonstrated its practicality and safety in a large group of selected patients (5). To expose the adrenal gland in the retroperitoneum, several trocars are typically required to retract adjoining visceral organs in traditional laparoscopic adrenalectomies. The posterior retroperitoneoscopic technique is conceived to be a more technically challenging but favourable method of adrenal surgery by granting direct access to the retroperitoneal organs. By decreasing the average operative time, the potential for perioperative haemodynamic fluctuations is reduced, which can restrict blood loss. Notably, this technique may be advantageous in MEN2A patients where 50–60% of phaeochromocytomas occur bilaterally (6).
Our patient expressed the struggle in taking daily supplementary medications as per the guidelines (7). Surgery in the future may be able to prevent the need for lifelong corticosteroid therapy. Cortical-sparing adrenalectomy appears to be a more viable treatment alternative to total adrenalectomy, in patients with hereditary phaeochromocytoma. Published threshold data require one-third of each gland to be preserved to maintain cortical function (8). Phaeochromocytoma recurrence after adrenalectomy is surprisingly not uncommon and occurs in roughly 20% of cases (9). Accordingly, patients with bilateral phaeochromocytoma undergo a unilateral cortical-sparing procedure with the entirety of the contralateral gland being removed. Theoretically, this halves the risk of recurrent phaeochromocytoma by preserving the adrenal cortex in only one side, as compared to both sides. A study of 89 patients undergoing cortical-sparing adrenalectomy, resulted in 60 patients (67%) progressing to corticosteroid independence and no recurrent diseases in 2 years (10). It is thereby recommended as treatment of choice due to the reassuring success rates and cortisone-free therapy in most of its cases.
CONCLUSION
Our case demonstrated the importance of genetic testing to effectively aid in diagnosing MEN2A, and the utility of MIBG in treating phaeochromocytoma. This report detailed the advancement of innovative operative techniques to improve care in MEN2A patients with phaeochromocytoma. Additional large-scale research is needed to gauge the pertinence of these new treatments in real world clinical scenarios.

{kind=link}