





Abstract
Mesenchymal liver hamartomas are benign tumors that can cause life-threatening abdominal distension and carry a risk for malignant transformation. In this case report, we describe a 13-month-old male with Beckwith–Wiedemann Syndrome (BWS) who presented with multiple mesenchymal liver hamartomas causing severe intra-abdominal mass effect. Imaging revealed six large multi-locular cystic lesions, ranging from 3.8 to 8.9 cm in diameter. The large size and spread of the tumors necessitated liver transplantation for complete removal. The patient successfully underwent cadaveric piggyback liver transplantation at 25 months of age. He was alive at 16-month follow-up without evidence of tumor recurrence or graft rejection. Histological examination of the hepatic masses revealed mucinous epithelial lining and abundant hepatocytes in varying stages of differentiation, supporting the diagnosis of mesenchymal hamartoma. To the best of our knowledge, this is the first reported case of liver transplantation in a patient with BWS as definitive treatment for unresectable mesenchymal liver hamartoma.
INTRODUCTION
Beckwith–Wiedemann Syndrome (BWS) is an overgrowth syndrome associated with a constellation of clinical findings, including macroglossia, macrosomia, umbilical abnormalities and craniofacial dysmorphia. Children with BWS are predisposed to malignant and benign tumors with reported cancer frequency ranging from 4.5 to 7% [1]. While most commonly reported tumors associated with BWS include hepatoblastoma and Wilms tumor, mesenchymal hamartoma of the liver (MLH) has been reported in patients with BWS [2]. Despite being classified as a benign tumor, if left untreated, MLH can grow dramatically, causing fatal compression of adjacent structures or destruction of functional hepatic parenchyma. Definitive treatment requires resection, and while transplant has been cited as a treatment option, only a few reported cases have required full organ resection and transplantation [3–6]. We report a rare case of unresectable MLH requiring liver transplantation in a patient with BWS.
CASE REPORT
A 13-month-old male with BWS and Kleinfelter Syndrome was referred to our institution for evaluation of asymptomatic liver masses. Hepatic workup completed at both 4 and 7 months of age showed normal liver function tests, but elevated alpha-fetoprotein (AFP) levels with a maximum of 167 000 ng/mL. MRI of the abdomen revealed six multi-locular cystic hepatic masses, ranging from 3.8 to 8.9 cm in maximal diameter, with corresponding hepatomegaly (19 cm) and significant intra-abdominal mass effect causing displacement of the proximal duodenum and proximal pancreas to the left of midline (Fig. 1). These imaging findings were consistent with the diagnosis of MLH.
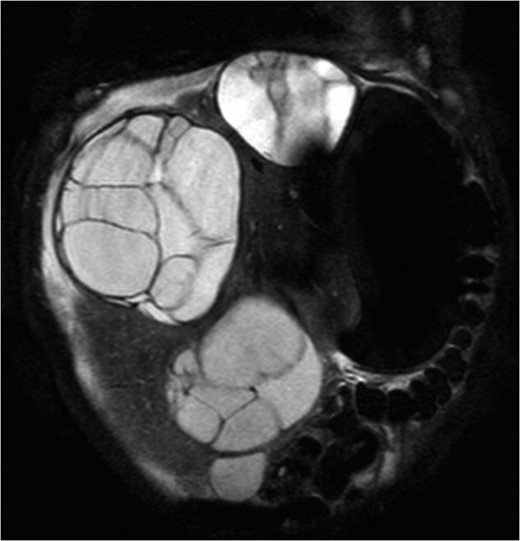
Coronal T2 abdominal MRI demonstrating multiple, large, multi-loculated cystic hepatic masses and significant hepatomegaly.
Due to risk of continued growth of the hamartomas, treatment with surgical resection was necessitated. The extensive hepatic involvement with multiple lesions scattered through the liver precluded partial resection; complete resection with liver transplantation was, therefore, deemed the only option. The patient underwent a cadaveric piggyback whole liver transplant with choledochocholedochostomy at 25 months of age. The patient tolerated the procedure well and was successfully extubated on postoperative Day 1.
The postoperative course was complicated by mild acute cellular rejection with correlating central venulitis, periheptocyte necrosis, and mixed cellular infiltrate seen on transjugular liver biopsy, which was treated with intravenous corticosteroid bolus and increased tacrolimus. The patient was discharged from the hospital on postoperative Day 42 in good condition. Serum AFP returned to normal levels by 2 months post-transplant, and histological findings of acute cellular rejection were largely resolved 3 months later. The patient was alive at 16-month follow-up without evidence of tumor recurrence or graft rejection.
Surgical pathology of the native organ revealed a 1338.6 g liver mildly distorted by soft to rubbery nodules extending to the intact capsule. Within the parenchyma were six well-delineated, white-tan, multi-loculated cystic masses. The contained fluid was white-tan and cloudy with watery to mucinous consistency. Histological examination of the cysts revealed mucinous epithelium in the lining and loose, edematous, myxoid to sclerotic tissue within the stroma. Abundant hepatocytes in varying stages of differentiation appeared in many configurations along the periphery of the masses, with more mature hepatocytes demonstrating ductal or cholangiolar differentiation. Areas of myxoid stroma with abnormal, branching bile ducts were seen focally. These histological findings were consistent with the diagnosis of mesenchymal hamartoma.
DISCUSSION
While patients with BWS are predisposed to development of several embryonal tumors [1, 7], MLH in these patients is rare [2]. Definitive treatment of MLH consists of complete resection, as untreated disease may result in fatal abdominal distention [8] or progression to undifferentiated, malignant sarcoma. More conservative approaches with partial resection, cyst drainage or cyst marsupialization have been linked to complications, including intractable ascites, liver abscesses, benign or malignant recurrence, and poor survival. A review of 11 incomplete resections resulted in only two patients alive at 1 to 17 year follow-up [9]. While MLH tumors may spontaneously regress, complete resection is still indicated as progression to malignancy has been reported even after 50% size reduction [10]. Thus, large or difficult-to-resect tumors present a challenge in the management of MLH patients. Sequential resection can be considered in certain cases of massive tumors [10], but unresectable tumors may necessitate liver transplantation. Five cases of MLH treated with liver transplantation have been previously reported in the literature—three infants and two adults, with four successful outcomes and one death due to intraoperative hemorrhagic shock [3–6]. To the best of our knowledge, this is the first report in the literature of liver transplantation in a patient with BWS for the treatment of unresectable MLH. While MLH may have been incidental in this patient, the management of tumors occurring in early life is of great importance in patients with BWS, as the majority of malignancy seen in children with BWS occurs in the first 10 years of life [7].
In conclusion, patients with BWS are at increased risk for embryonal tumors, most commonly hepatoblastoma and Wilms tumor. We present mesenchymal hamartoma as a rare tumor in this population. Complete resection of the tumor is advised in the management of all mesenchymal hamartomas. In cases of unresectable MLH, liver transplantation may be required as definitive treatment due to the morbidity and mortality associated with continued growth or progression to malignancy of hamartomas with incomplete resection. We report the first successful case of liver transplantation as definitive treatment for mesenchymal liver hamartoma in a patient with BWS.
CONFLICT OF INTEREST STATEMENT
None declared.

{kind=link}