





Abstract
Vascular anomalies constitute some of the most difficult diagnostic and therapeutic enigmas in the practice of medicine, ranging from an asymptomatic birthmark to life-threatening congestive heart failure. Hemangiolymphangiomas (HLA) are extremely rare vascular malformations of both lymphatic and blood vessels in which 80–90% are diagnosed during the first 2 years of life. Though rare, these vascular malformations have promising clinical outcomes. We report a case of a 28-year-old male who presented with a neck mass of unknown etiology. A computed axial tomography scan revealed a cystic mass, and subsequent aspiration biopsy showed lymphatic fluid. The mass was surgically excised and identified as a 6 × 6 × 3 cm3 multilocular cystic mass weighing 81 g. Histopathology showed cellular characteristics consistent with HLA. We review the salient clinical and pathophysiologic features of vascular anomalies.
INTRODUCTION
Hemangiolymphangiomas (HLA) is an extremely rare vascular malformation comprised of both endothelial and lymphatic components. It is a subgroup under the umbrella of endothelial malformations. Historically, endothelial malformations were named per size of channels and type of fluid contained in the lesion. They were classified as capillary hemangiomas, strawberry hemangiomas and cavernous hemangiomas. Lymphatic lesions were referred to as lymphangiomas or cystic hygromas. This classification was replaced by Mullikin and Glowacki [1] who classified lesions into two large groups, namely, hemangiomas and vascular malformations. This new classification was based off the natural history, cellular turnover and histology.
According to literature, 40–60% of HLAs are discovered at birth, 80–90% during the first 2 years of life, and decreases in frequency with age. The risk of developing HLA is greater in premature babies and in live newborns, incidence of 1:12 000 [2]. The most common site of presentation occurs in the anterior and posterior cervical triangle of the neck. HLAs have also been reported to occur in the duodenum, oral and maxillofacial region, colon, bladder, testis and vertebral column [3–5]. Here in we present a 29-year-old male with a persistent neck mass diagnosed as HLA.
CASE
Our patient was a 29-year-old Caucasian male who presented to the clinic with a non-painful right sided anterior neck mass. Aspiration of the mass yielded 20 ml of straw colored fluid and resulted in a reduction of the mass’s size. Cytology of the fluid was negative for any malignant cells, and confirmed to be normal lymphatic fluid. The patient was seen 10 days later when the mass returned to its previous size. Computer tomography (CT) scan of the neck revealed an ovoid cystic appearing lesion deep to the sternocleidomastoid muscle, adjacent to the carotid artery. It measured ~7.6 cm in length by 6.8 cm in transverse dimensions and by 3.9 cm in AP dimensions. No associated lymphadenopathy was noted. However, the internal jugular vein was medially displaced (Fig. 1).
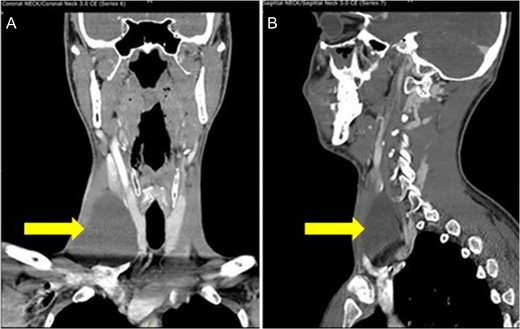
CT scan of the neck shows an ovoid cystic appearing lesion present at the base of the right neck (A: Coronal view and B: sagittal view).
One week after imaging, a right neck dissection was performed without complication. The procedure resected a 6 × 6 × 3 cm3 multiloculated cystic mass containing bloody fluid and weighed ~81 g. The specimen was preserved in formalin and sent to pathology. Histopathology showed dilated lymphovascular spaces with numerous red cells in the lumen and lymphoid aggregates in the cyst wall. Immunohistochemical staining with CD31 and D2-40 highlighted an endothelial cellular lining in one section of the cyst (Fig. 2). Based on these results, the diagnosis of HLA was confirmed.
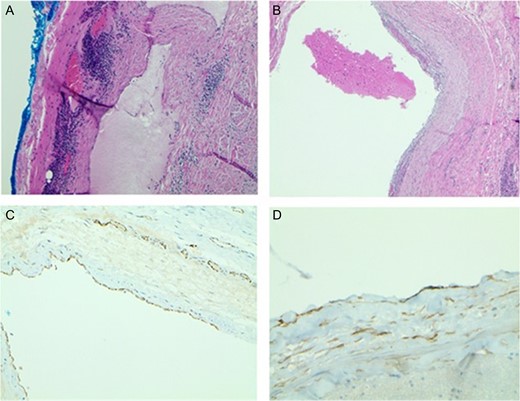
(A) Light Microscopy view of cyst wall with lymphoid aggregates (×20). (B) Blood in the dilated lymphovascular space (×40). (C) CD31 highlights the lining of endothelial cells (×40). (D) D2-40 positive stain highlights the lining of endothelial cells (×100).
DISCUSSION
While many classifications of vascular anomalies exist, they can be simply divided into hemangiomas and vascular malformations. It is pertinent to accurately diagnosis vascular anomalies as they possess different options for management.
Hemangioma
While the etiology of hemangiomas is not well understood, several risk factors have been reported in the literature. A familial history of hemangioma has been reported in 12% of cases, as well as the maternal use of fertility drugs, use of erythropoietin and a breech birth [6–9]. The incidence of hemangioma increases in preterm infants weighing <1 g [10]. Other prenatal risk factors include maternal chorionic villus sampling or amniocentesis, older maternal age, multiple gestation pregnancy, placenta previa and pre-eclampsia [7, 11, 12]. Placental anomalies including retroplacental hematoma, infraction and dilated vascular communications have been implicated in the development of hemangiomas, all of which are associated with placental hypoxia [13].
Several theories have been postulated to explain the pathogenesis of hemangiomas. Harbi et al. [14] report that hemangiomas may be due to dysregulated stem cells that remain in an immature arrested stage of development. This is in line with the findings of Boye et al. [15] who reported that endothelial cells from proliferating hemangiomas are a result of clonal expansion, and that hemangioma-derived cells differ from normal endothelial cells in their rates of proliferation and migration. Furthermore, the migration of hemangioma endothelial cells was stimulated by the angiogenesis inhibitor endostatin, in contrast to inhibition which is seen with normal endothelial cells [15]. This phenomenon suggests the possibility of an altered cellular phenotype. Evidence of mutagenesis in hemangiomas has been shown in loss of heterozygosity mutations on chromosome 5q [16]. Additionally, somatic missense mutation in mitogen-activated protein kinase kinase kinase 3 (MAP3K3) has been implicated in vascular malformations [17].
The expression of placental genes in hemangiomas suggests an alternative theory. Hemangiomas display high levels of GLUT1 glucose transporter which is present during embryonic development, but subsequently lost in most mature tissues except at the blood–tissue barriers [18]. Other placental antigens have been found in hemangiomas including FcγRII, Lewis Y antigen and merosin [19]. This theory is attractive as it accounts for the exclusive congenital presentation of hemangiomas [20].
Hemangioma proliferation appears to be related to an imbalance between positive and negative angiogenic factors expressed by the neoplasm and adjacent normal tissues [21]. This imbalance is supported by the overexpression of basic fibroblast growth factor (bFGF), vascular endothelial growth factor (VEGF), proliferating cell nuclear antigen and type IV collagenase in proliferating tumors [21–23]. Additionally, vascular endothelial growth factor receptor 1 (VEGFR1) is suppressed with VEGFR2 constitutive signaling. Mutations in VEGFR2 or the integrin-like receptor TEM-8 inhibit nuclear factor of activated T cells (NFAT) which leads to VEGFR1 suppression [24].
The goals of treating hemangiomas are to prevent loss of function and minimize scarring. Therapeutic options include the use of cryotherapy, radiation therapy, steroids, interferon alfa-2a, laser, embolization, sclerosing agents, antifibrinolytics, systemic propranolol and surgery for larger lesions. While the mechanism of action for corticosteroid use is unclear, steroids are known to inhibit angiogenesis and induce capillary regression [25]. However, the long-term use of steroids increases the risks of severe side effects and should be discontinued if no response is seen within a week after initiating therapy [26]. Interferon alfa-2a has gained attention due to its reported ability to reverse pulmonary hemangiomas, life-threatening hemangiomas and diffuse neonatal hemangiomas [27]. Interferons inhibit angiogenesis and stimulate endothelial cell prostacyclin formation, thereby preventing platelet trapping [28]. However, due to severe neurological complications, interferons are not recommended.
Beta blockers have been observed to inhibit the proliferation of hemangiomas, and have become first line medical therapy by many clinicians [29]. In contrast to the side effect profile of corticosteroids, beta blockers represent a safer and more effective means to treating these lesions. A randomized control trial involving 460 patients found that propranolol exhibited a 60% rate of successful treatment (complete or nearly complete resolution of the hemangioma), compared with a 4% rate among patients treated with placebo [30]. While the mechanism of action is unclear, proposed mechanisms include vasoconstriction, inhibition of angiogenesis, regulating renin–angiotensin system, inhibition of nitric oxide production and the stimulation of apoptosis [31]. Other medical therapies including vincristine, imiquimod and antiangiogenic agents such as VEGF or bFGF antagonists have efficacy in treating hemangiomas, however, their side effect profile limits them from being of clinical value [31]. While there are several laser therapies available, flash-lamp pumped pulsed dye laser (FPDL) is the treatment of choice for superficial hemangiomas, with a 60% response rate [26, 32]. The use of surgery in the management of hemangiomas depends on location, depth of invasion, age and cosmetic considerations.
Vascular malformation
Vascular malformations are classified by the vessels present, such as arterial, arteriovenous, venous, capillary or lymphatic malformations. Mixed vessels encompass congenital lymphangiomas or lymphatic anomalies which are aggregates of lymph vessels [33]. When lymphangiomas are filled with blood, they are referred as a mixed hemangiolymphangioma, which is an extremely rare entity. Unlike hemangiomas which proliferate, vascular malformations enlarge in proportion to the growth of the individual and expand by hypertrophy [34]. Although they are not clinically evident at birth, they may present at later stages of life, becoming more evident following trauma, infection or pregnancy [35]. While these tumors are benign, they have a tendency to invade underlying tissues, and may recur locally [33].
Vascular malformations can be further characterized as either high-flow (arterial components) or low-flow (venous and lymphatic components) [5, 36]. High-flow lesions are firm on palpation, while low-flow are soft and compressible.
Recognizing a lesion as high-flow is clinically important as it is associated with congestive heart failure, embolism, bleeding and ulceration [36]. The clinical presentation is further complicated as HLAs are associated with a multitude of syndromes such as Rendu–Osler–Weber syndrome, Sturge–Weber–Dimitri syndrome, blue rubber bleb nevus syndrome, Parkes–Weber syndrome, Bannayan’s syndrome, Sturge–Weber–Krabbe syndrome, Klippel Trenaunay syndrome, Servelle–Martorell syndrome, Maffucci’s syndrome and von Hippel–Lindau syndrome [37–41].
In addition to a thorough history, CT and MRI have been successfully used to diagnose soft tissue lesions [42]. The diagnosis of vascular malformation is confirmed using immunohistopathology which detects positive CD31 and D2-40 reactivity. Interestingly, new markers for detecting abberant lymphatic vessels using Prox-1 and VEGFR3 antibodies have been reported in the literature [43].
Vascular malformations can be managed by laser therapy, embolization, or surgical excision. The treatment option is dependent on the depth of vascular malformation and anatomical site. Low-flow capillary and venous malformations can be successfully treated by interstitial laser therapy. When laser therapy is used, more than one session is required to achieve complete vascular involution. Lymphatic malformations are treated surgically, or if access is challenging, sclerotherapy is alternatively used [44]. High-flow malformations are treated by embolization with or without surgery. Embolization aims to occlude the nidus of the malformation. Unlike hemangiomas, surgical resection of vascular malformations is recommended to prevent recurrence. Prior to operation, patients must be assessed by ultrasound and radiographic imaging (MRI or CT) to thoroughly delineate the extent of the lesion [45].
Clinicians should have a high index of suspicion as HLAs may mimic other lesions on presentation, radiographically, and histologically. The differential diagnosis of a neck mass is extensive, and covers both benign and malignant etiologies. As a result, the differential diagnosis includes congenital (branchial cleft cyst, thyroglossal duct cyst—most common, vascular anomalies, laryngocele, ranula, thymic cyst, etc.), inflammatory (infectious and noninfectious disorders), and neoplastic (squamous cell carcinoma—most common, lipoma, thyroid masses, etc.) [46]. This list is not exhaustive, but it does paint the picture that the differential of neck masses encompasses a broad spectrum of etiologies.
CONCLUSION
Vascular anomalies are characterized as hemangiomas or vascular malformations. Accurate diagnosis is imperative for appropriate treatment. HLA is a vascular tumor of both lymphatic and blood components. We report a very rare case of HLA in the neck of an adult male which was treated by means of surgical excision with a favorable clinical outcome. While HLA is benign, they can cause bodily distortion due to local growth and invasion of surrounding structures. Furthermore, we would like to highlight that the evaluation of neck masses encompasses congenital, inflammatory and neoplastic etiologies. To this end, neck masses require diagnostic vigilance and should be immediately evaluated.
CONFLICT OF INTEREST STATEMENT
None declared.

{kind=link}
{kind=link}