





Abstract
Opitz syndrome is a rare genetic disorder which has been well defined; however, the surgical treatment of the anomalies has not been codified. The objective is to review the literature and describe the surgical priorities in the treatment of Opitz syndrome. This report is unique in the fact that it describes a surgical approach to the treatment of the deformities. Better outcomes are achieved with preoperative analysis of the deformities and surgical planning. Simultaneous soft tissues and bony reconstruction with grafts can achieve long lasting results and decrease recurrence rates.
INTRODUCTION
Opitz G/BBB Syndrome is a constellation of congenital deformities including laryngotracheoesophageal anomalies, developmental delays, genital defects as well as craniofacial defects. The syndrome receives its name from a combination of the names of the first families affected as well as the doctor who first recognized and described the pattern consistent with the syndrome [1, 2].
The syndrome is rare and manifested through two distinct inheritance patterns. It can be inherited through an X-linked arrangement, known as XLOS, or through an autosomal dominant pattern. In a small subset of patients, no recognizable inheritance pattern is appreciated [3, 4].
The incidence of XLOS is ~1 in 10 000–50 000 and affects males with females being carriers. It is likely that the incidence is higher due to lack of recognition leading to underdiagnosis. The abnormality responsible for this syndrome is a mutation in the MID1 gene which produces the midline-1 protein responsible for microtubule binding. The normal function of the protein allows for activation of key elements of the cytoskeleton as well as induction of cell migration. Therefore, mutations in this gene result in defects of mitosis through cell division and migration of cellular elements [5].
Autosomal dominant Opitz is part of larger syndrome referred to as 22q11.2 Deletion Syndrome. It is estimated to affect ~1 in 4000 individuals. The exact genes involved in clinical signs and symptoms of Opitz G/BBB syndrome is not known, while in others it is caused by a mutation involving the SPECC1L gene. It is important to note that the area of deletion is not typically seen in individuals with autosomal dominant Opitz syndrome or the 22q11.2 deletion syndrome. This gene is responsible for the production of cytospin-A which interacts with cytoskeletal elements and microtubule stabilization. This is important in cell migration of facial features. This is likely why individuals with this mutation have cleft lips and palates [1, 6–8].
The clinical manifestations of the syndromes typically present in similar manners independent from the inheritance pattern.
Patients with laryngotracheoesophageal defects such as laryngotracheal diastema can present with recurrent respiratory infections, pneumonia, dysphagia and possibly require tracheostomy [3]. Developmental delays or intellectual disabilities occur in approximately half of the patients. They may also present with neuropsychiatric disorders such as delayed motor skills caused by agenesis of the corpus callosum and cerebellar vermis or symptoms consistent within the autism spectrum [9].
Approximately 85% of males affected with the disorder may present with genital deformities such as hypospadias, a bifid scrotum or cryptorchidism [1].
Other less common presenting deformities can be imperforate or ectopic anus and congenital heart defects such as ventricular septal defects [3].
Craniofacial defects will be manifested as ocular hypertelorism, prominent forehead and widow's peak, cleft lip and palate, broad nasal bridge, anteverted nares, encephalocele and bilateral low-set prominent ears. These defects may only be present in 50% of affected individuals and also vary between affected family members [3].
CASE REPORT
Our case is a 21-year-old male born with clinical manifestations consistent with Opitz G/BBB syndrome including severe bilateral cleft lip and palate, hypertelorism, low-set prominent ears and midfacial hypoplasia. His history was complicated with respiratory distress requiring tracheostomy at 8 months of age. He had also developed gastric volvulus requiring partial gastric resection and gastrostomy.
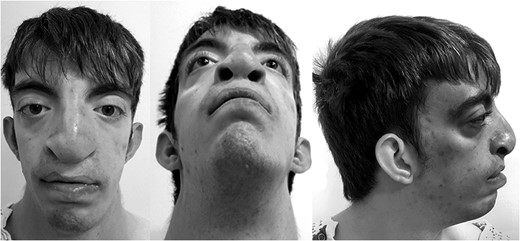
Preoperative evaluation showing bilateral cleft lip and palate, hypertelorism, low-set prominent ears and midfacial hypoplasia.
Functional evaluation of the nose revealed a positive Cottle maneuver for bilateral internal valve collapse, external valve collapse on forced inspiration, bilateral retracted and notched ala, shortened columella and poor tip projection.
Bilateral internal nasal valve collapse with bilateral spreader grafts
Bilateral external nasal valve collapse with bilateral batten grafts
Bilateral retracted and notched ala with bilateral rim grafts
Shortened columella with double V–Y flaps
Poor tip projection with columellar strut
Hypoplastic midface and pyriform rim with costocondral rib grafts
Lip shortening and vermillion deficiency with dermal graft
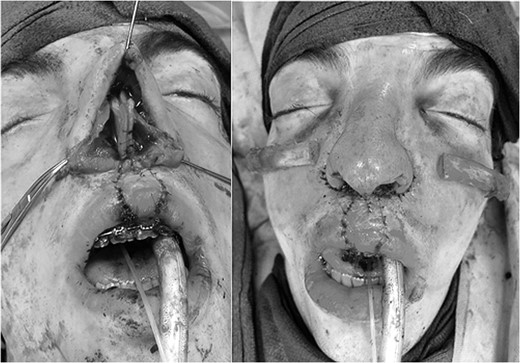
Intraoperative findings with correction of cleft lip and rhinoplasty.
A 6-month follow-up was scheduled to review the restorative results. Surgical correction of the severe cleft lip, nasal deformities and midfacial hypoplasia showed significant improvement in symmetry and aesthetic result. Nasal airway function was optimal at both the internal and external nasal valves as evidenced by negative Cottle maneuver. The alar base was corrected to a symmetric position. The grafts maintained their structure and had little resorption. Bone grafts proved to provide adequate scaffolds at the pyriform rim. The patient has not undergone further revision.
DISCUSSION
A thorough search through the literature reveals that the majority of available data published on Opitz Syndrome only defined the clinical manifestations of the syndrome. There is little published data regarding the surgical management.
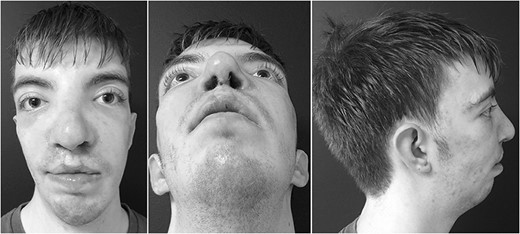
Postoperative results.
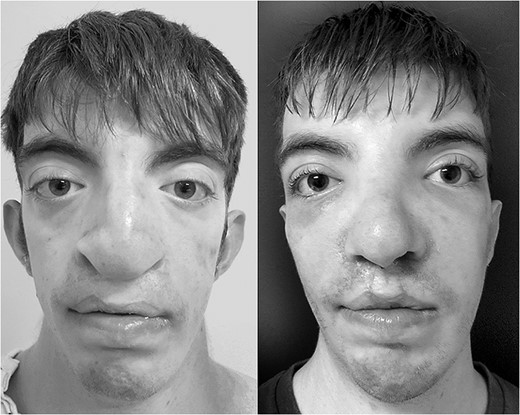
Side by side comparison of preoperative findings and postoperative results.
Our report highlights a functional and rational approach to correct the craniofacial deformities associated with Opitz G/BBB Syndrome. It provides an enhancement in anatomic function which can decrease diseases associated with defects. It also produces a considerable improvement is aesthetic outcomes.
CONFLICT OF INTEREST STATEMENT
None declared.
FUNDING
The authors certify that they have no affiliations with or involvement in any organization or entity with any financial interest or non-financial interest in the subject matter or materials discussed in this manuscript.

{kind=link}
{kind=link}
{kind=link}