





Abstract
Alveolar soft part sarcoma (ASPS) is a very rare sarcoma, report <1% of all soft tissue tumor. Majority of cases were young adult and tumor occurred in lower extremities and trunk. Here, we present a case of 53-year-old Thai female with rapidly glowing mass over her right forearm. The magnetic resonance imaging scan demonstrated a hypervascular mass with multiple feeding vessels located in flexure tendon of right forearm. Ultrasound-guided biopsy revealed malignant poorly differentiated epithelioid neoplasm with clear cell feature and focal necrosis. Surgery may be considered the first-line treatment in localized ASPS and may potentially increase long-term survival. Complete surgical excision is the mainstay of treatment. TFE3 and Cathepsin K immunohistochemistry are useful in confirming a diagnosis of ASPS with a distinctive clinicopathologic features.
INTRODUCTION
Alveolar soft part sarcoma (ASPS) is a very rare sarcoma, report <1% of all soft tissue tumor. Majority of cases were young adult and tumor occurred in lower extremities and trunk. Our case is rare case that occurred in upper extremity and our patient age is oldest in the English literature. No guideline of treatment due to rare has been reported. We report our treatment and short-term result. To best of our knowledge, this rarely report.
CASE REPORT
A 53-year-old Thai woman presented with a mass at right forearm rapidly growing for 5 months (Fig. 1). She had no pain and had normal forearm and hand function. She had no skin and sensory involvement. On physical examination, there is a firm mass over right forearm measuring ~8 cm in diameter. No notable grossly skin involvement and the mass appears not fixed to bony structure. No pain or sensory deficit is identified.
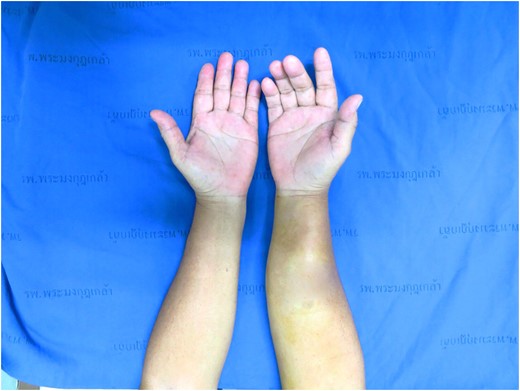
Preoperative picture, tumor located over right forearm.
The magnetic resonance imaging (MRI) scan demonstrated a hypervascular mass with multiple feeding vessels located in flexure tendon of right forearm (Fig. 2). Ultrasound-guided biopsy revealed malignant poorly differentiated epithelioid neoplasm with clear cell feature and focal necrosis (Fig. 3). Workup distant metastasis prior operation chest X-ray and intra abdominal were cleared. Bone scan study shows the areas of increased radiotracer accumulation at right seventh of right rib posterolaterallly, L2, right sacroiliac region, proximal part of left humerus and proximal of right femur, multiple foci of osseous metastasis cannot be rule out.
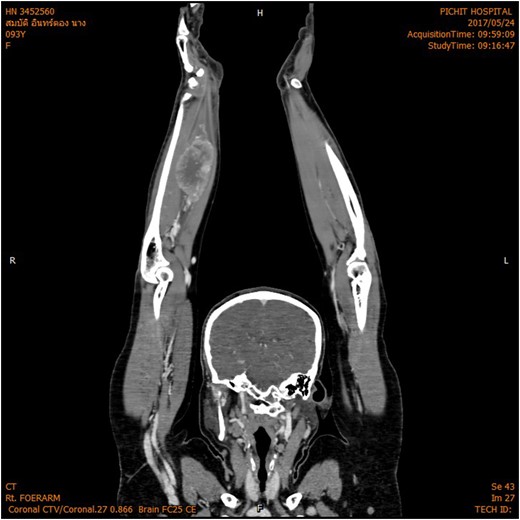
MRI shown hypervascular mass with multiple feeding vessels located in flexure tendon of right forearm.
Ultrasound-guided biopsy revealed epithelioid cell neoplasm with clear cell features.
Under operation, patient was positioned supine right arm extend over arm rest. Under general anesthesia, lazy-S incision was performed, dissected and identified tumor. Tumor involved and confined in deep group of flexor tendon of right hand (flexor digitorum profunda), no bony involvement. En bloc resection with 1 cm margins was performed (Fig. 4).
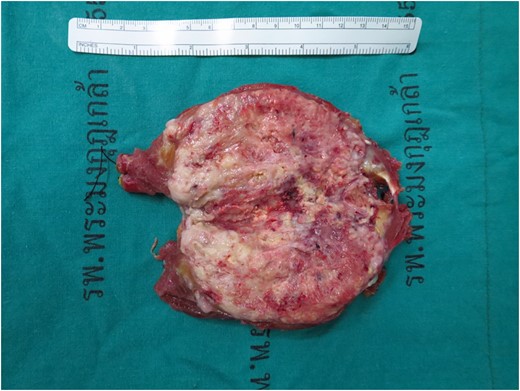
Gross specimen, tumor size 11.5 cm × 5.0 cm × 5.0 cm, a poorly circumscribed, large gray–yellow mass with semi-soft tan homogenous cut surface with large areas of hemorrhage and necrosis.
The specimen consists of an 11.5 cm × 5.0 cm × 5.0 cm ovoid-shaped fragment of pink–red soft tissue with orienting sutures present. Skeletal muscle is present at one margin and the opposite margin is smooth pink–yellow and glistening. Sectioning reveals a poorly circumscribed, large gray–yellow mass with semi-soft tan homogenous cut surface with large areas of hemorrhage and necrosis of nonspecific appearance (Fig. 4). The mass measures ~8.5 cm × 4.8 cm and grossly abuts all surgical margins with a thin capsule separating it from the resection margins.
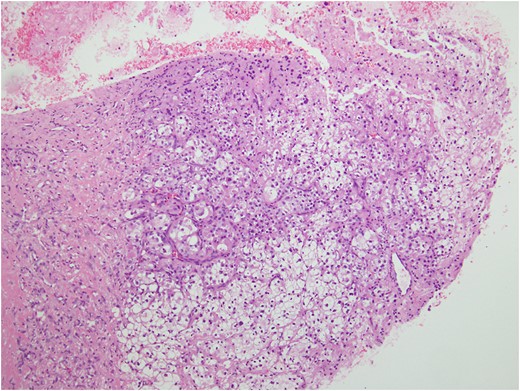
Histologic sections of the mass show distinct variably sized and shaped collections of uniform polygonal tumor cells, separated by fibrovascular septa and delicate capillary-sized vascular channels lined by flattened endothelium at low magnification (Fig. 5). Within the tumor nests, there is loss of cellular cohesion and necrosis of the centrally located cells in the nests results in the pseudo-alveolar pattern (Fig. 6). Vascular invasion is also present (Fig. 7). The individual tumor cells have distinct cell borders and abundant eosinophilic to clear, somewhat granular cytoplasm surrounding a central nucleus with variably sized nucleolus. Nuclear atypia is rarely seen. Mitotic figures are uncommon. The cells contain rhomboid or rod-shaped crystalline inclusions that are faintly apparent on routine histology and are better demonstrated with periodic acid-Schiff stain after diastase digestion (Fig. 8).
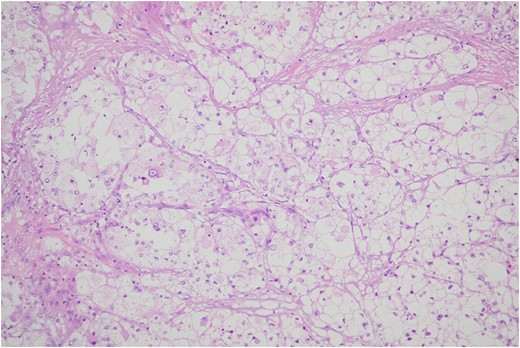
Tumor cells are arranged in organoid or nesting pattern. The nests are vary in size and shape and are separated by delicate thin fibrous septa containing vascular channels.
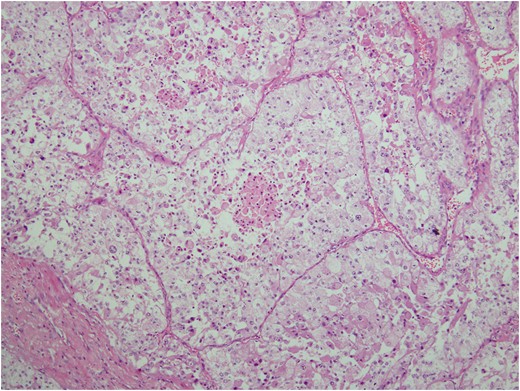
Loss of cellular cohesion and necrosis of the centrally located cells in the nests.
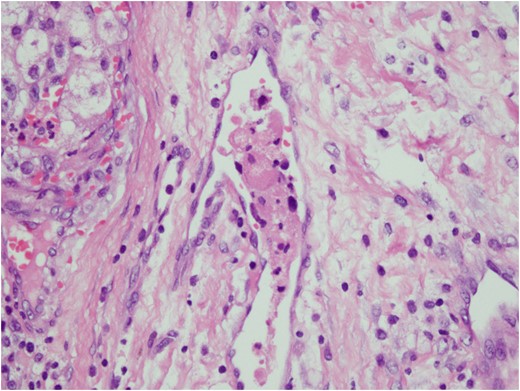
Vascular invasion by tumor cells.
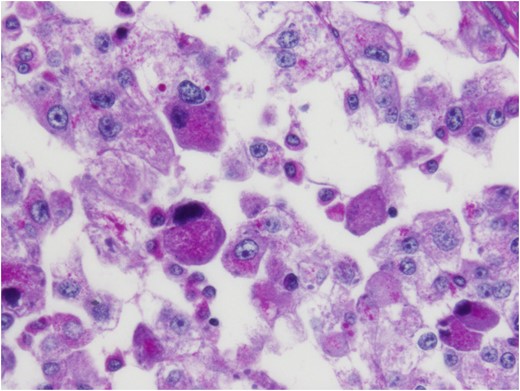
Crystalline inclusions demonstrated with periodic acid-Schiff stain after diastase digestion.
Extensive immunohistochemical studies are performed and most of the tumor cells show nuclear expression of TFE3 (Fig. 9). Immunoreactivity for Vimentin outlines the delicate vascular envelope around group of tumor cells and some of tumor cells are positive (Fig. 10). Immunostaining for Desmin, MyoD1, Myogenin, S100, EMA and AE1/AE3 are consistently negative.
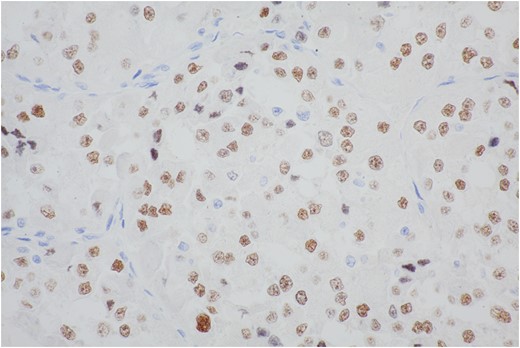
The majority of cells show moderate to strong nuclear staining with the antibody to the carboxy-terminal portion of TFE3 retained in the fusion protein.
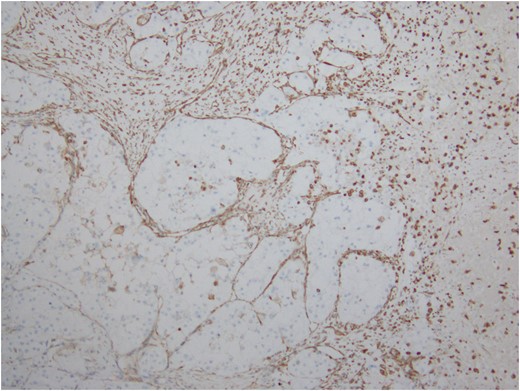
Immunostain for Vimentin outlines the delicate vascular envelope around group of tumor cells.
DISCUSSION
ASPS is a very rare sarcoma, report <1% of all soft tissue tumor [1]. ASPS was first described by Christopherson et al. [2] at Memorial Sloan Kettering Cancer Center, NY, USA.
A diagnosis is made by a combination of histology and immunohistochemistry; looking for strong nuclear expression of transcription factor. Predominantly affects young adolescents, higher incidence among patients between 15 and 35 years of age, but rare before age 5 and after age 50 years [3]. The disease has female predominance in the first three decades and reverses, thereafter, the reported female to male ratio is ~2 : 1.
Here, we report a 53-year-old female with ASPS. To the best of our knowledge, her age is the oldest patient of ASPS in the English literatures. ASPS is usually a slow-growing tumor; however, in our case, patient presented with a rapidly growing lesion over a period of 5 months, which has not been described previously. ASPS often occur primarily in the lower extremities and trunk, our case is a rare case that occurs in upper extremity. Other rare locations of ASPS were reported such as intracranial, tongue and larynx. ASPS is hypervascular soft tissue sarcoma. As seen in our case, MRI studies also show hypervascular lesion and have many feeding vessels into tumor.
Surgical resection, chemotherapy and radiotherapy have been employed individually and in combination in the management of primary ASPS. Given the rarity of the disease, reports into the outcomes of these treatment modalities are limited to large case series and do not address ASPS effecting one specific anatomical region. Surgery may be considered the first-line treatment in localized ASPS and may potentially increase long-term survival. Complete surgical excision is the mainstay of treatment. Role of adjuvant therapy remains controversial, but radiotherapy/chemotherapy may be given in cases of inadequate surgical excision [4].
Previous study by Li et al. [5], 14 patients achieved partial remission and 10 patients achieved stable disease. Median progression free survival was 41.0 months (95% confidence interval: 7.7–74.4 month). Median overall survival (OS) was not reached. The 1- and 4-year OS rates were 90.0% and 60.0%, respectively. The most common adverse events (AEs) were bleeding (35.7%), hair and skin color change (37.5%) and mucositis (28.6%)
In our case, we can achieve complete resection even cannot rule out bone metastasis. We decided to give our patient only postoperative radiotherapy and closely monitor progression of bony lesion by bone scan monthly for first 3 months.
The prognostic parameters of ASPS include age at diagnosis, tumor size and the presence of metastasis [1]. Given the age of our patient is 53 year old, the tumor size measures ~8 cm, metastasis cannot be ruled out and the tumor is completely resected, the prognosis should be classified at moderate risk because in old age group have better prognosis than younger age group. Tumor size was also in moderated in size.
ASPS has a distinctive and characteristic, nested or organoid growth pattern. The nests tend to be uniform in size and shape, although some variations may be present. The nests are separated by delicate sinusoidal vascular channels lined by a flattened, single layer of endothelial cells. The cells may appear discohesive with focal necrosis in the center of the nests giving rise to the so-called, commonly seen, pseudo-alveolar pattern [6].
The diagnosis in the excision specimen of our case is reasonably straightforward, given the presence of a classic alveolar pattern and the presence of periodic acid-Schiff-positive, diastase-resistant crystalline structures together with corroborating immunoreactivity for TFE3, an antibody directed against the C-terminus of the TFE3 which has emerged as a highly sensitive and specific marker of the ASPS [1, 7]. Since it has been discovered that ASPSs are characterized by a tumor-specific der(17)t(X;17) (p11;q25) that fuses the transcription factor 3 (TFE3) gene at Xp11 to the ASPL gene at 17q25, creating an ASPL–TFE3 fusion protein [8]. However, the diagnosis of ASPS can be problematic in a small tissue biopsy and the differential diagnosis of ASPS in this old age patient can be fairly broad and includes neoplasms that may show alveolar or organoid patterns of growth and cells with abundant clear-to-eosinophilic cytoplasm. Metastatic renal cell carcinomas, adrenal cortical carcinomas, hepatocellular carcinomas, perivascular epithelioid cell neoplasms (PEComa), granular cell tumors and paragangliomas may mimic ASPS by their abundant eosinophilic to clear cytoplasm. Furthermore, alveolar rhabdomyosarcomas can be also involved in the differential diagnosis, given their alveolar pattern. In most circumstances, immunophenotypic profile of the biopsied tumor including pertinent negatives as well as TFE3 expression should resolve the problem and help in this differential diagnosis [1]. On the other hand, nuclear expression of TFE3 can be seen in a variety of different tumors, most of which harbor TFE3 gene fusions, including ASPS, Xp11 translocation renal cell carcinoma, ‘melanotic’ Xp11 translocation renal cell carcinoma and a subset of PEComas and epithelioid hemangioendotheliomas [9]. In addition to TFE3, Cathepsin K is also consistently and diffusely positive in ASPS and can be helpful in the differential diagnosis of ASPS because it is negative in conventional RCC, ASPSCR1-TFE3 translocation RCC, adrenocortical carcinoma and paraganglioma. However, its role in differentiating ASPS from other potential mimickers is relatively limited because it is positive in melanoma, granular cell tumor, and other tumors [9]. TFE3 and Cathepsin K immunohistochemistry are therefore useful in confirming a diagnosis of ASPS with a distinctive clinicopathologic features.
CONFLICT OF INTEREST STATEMENT
None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}