





Abstract
Langerhans cell histiocytosis (LCH) is a rare histiocytic disorder of unpredictable clinical course and varied modes of presentation. The spectrum of presentation is wide, ranging from isolated eosinophilic granulomas to multiple lesions and diffuse systemic involvement. We present the case of a 52-year-old man, who presented with an 8-week history of worsening otalgia and superficial temporal tenderness attributed to otitis externa within the community and subsequently giant cell arteritis. Computed tomography and magnetic resonance imaging were undertaken due to atypical features, which demonstrated bony destruction within the right greater wing of the sphenoid, squamous part of temporal and mastoid bone, with middle cranial fossa communication. Intra-orbital extension was noted with abutment of the lateral rectus muscle. Mastoid biopsies demonstrated a mixture of lymphocytes, eosinophils and monomorphic epithelial cells with pale cytoplasm and focal areas of granulation tissue/necrosis. The features were consistent with a diagnosis of LCH, and the patient was subsequently transferred to a tertiary centre for definitive treatment.
Case History
We present a rare case of a 52-year-old man, who presented to our otolaryngology emergency clinic, with an 8-week history of worsening right-sided otalgia and superficial temporal tenderness attributed to otitis externa in the community. He had been commenced on several courses of oral antibiotics without any alleviation in his symptoms, and concerns were raised regarding the possibility of necrotising otitis externa given a history of diabetes mellitus. On review, there was no overt evidence of an infective pathology, and as a result, the patient was discharged accordingly. A fortnight later, the patient re-presented within the community and was subsequently referred to the medical team for assessment and management of a potential giant cell arteritis (GCA). A review was undertaken by the rheumatology team, who felt the nature of his presentation was atypical of GCA but was nevertheless commenced on steroid therapy, and a further review by the otolaryngology team was requested. On admission, with the exception of a mildly elevated erythrocyte sedimentation rate (49) mm/hr, all biochemical and haematological parameters were within normal range. On examination, pain was localised superficially to the right temporal artery distribution. Otoscopy was unremarkable, with no evidence of granulation tissue, polypoidal masses or discharge within the external auditory canal. On further questioning, the patient did report intermittent nocturnal deep rooted otalgia, in the absence of tinnitus or vertigo. This was coupled with mild intermittent right-sided retro-orbital pain within the preceding 4 months, which prompted radiological evaluation.
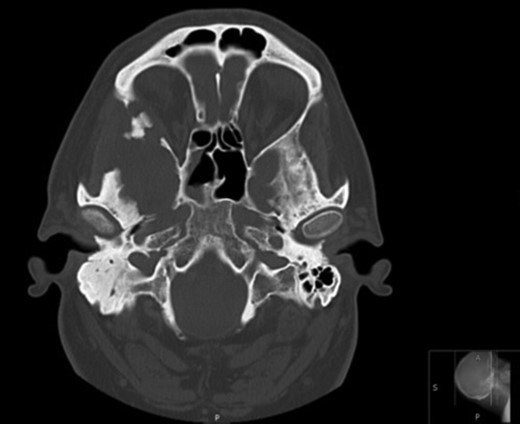
Axial CT images demonstrating bony involvement of the right mastoid and greater wing of the sphenoid.
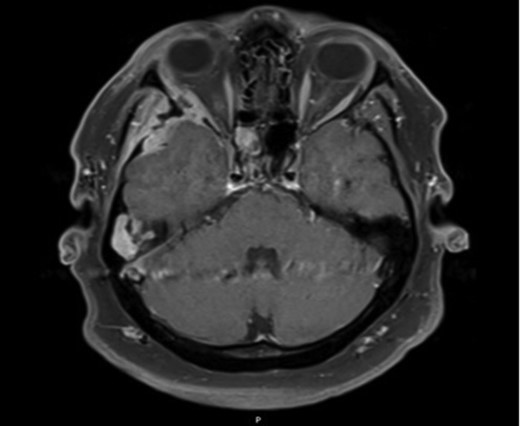
Axial MRI image demonstrating ocular involvement, with abutment of right lateral rectus muscle, as well as enhancement of temporal lobe.
Due to the extensive nature of the findings, a magnetic resonance imaging (MRI) scan of the head was undertaken and demonstrated bony erosion of the lateral wall of the right orbit, right superior rectus muscle abutment and minimal involvement of the antero-inferior surface of the temporal lobe. The appearances were suggestive of fibrous dysplasia, chronic inflammatory/infective pathology or less likely metastatic carcinoma.
A right cortical mastoidectomy was undertaken shortly after admission, and histopathological evaluation demonstrated areas with a mixture of lymphocytes, eosinophils and monomorphic epithelial cells with pale cytoplasms. Focal areas of granulation as well as necrosis were also noted. The epithelial cells stained with CD1a, S100 and CD68 indicating that they are epithelial cells. CD1a and CD68 show ghost cells in the necrotic areas, features consistent with a diagnosis of Langerhans cell histiocytosis (LCH).
The patient was subsequently referred to a tertiary centre for discussion at a regional skull base multidisciplinary meeting, and given the extensive nature of the disease process, surgical intervention was deemed futile, and an onward referral was made to haematology. A diagnosis of cranial diabetes insipidus, a finding in up to 30% of patients [1], was coincidentally made, and the patient was initially commenced on azathioprine (a single system regime). At a 2-month follow-up, a repeat MRI scan demonstrated drastic regression in the disease process, particularly within the temporal and suborbital components. Although the extensive involvement of >1 facial/cranial bones would categorise this process as multi-system LCH (thus warranting treatment with cytarabine and etoposide), adequate control of symptoms, coupled with radiological recovery, has dictated an observational approach with no further change to his current treatment regime.
Discussion
LCH (formerly referred to as Histiocytosis X), includes the disorders eosinophilic granuloma, Letterer–Siwe disease and Hand–Schüller–Christian disease. Although the aetiology remains uncertain, it is postulated to be a reactive disease secondary to abnormal immune regulation and demonstrates a predilection for the paediatric population [2–4]. Extensive skull base involvement at presentation is extremely rare [5] and usually confined to the orbit and cranial base [2, 6]
Otologic manifestations, although rare, may include aural discharge, postauricular swelling, vertigo and subjective hearing loss [2, 3, 6], which explains why in its early stages, the disease is attributed to an underlying acute or chronic inflammatory process [6]. Clinical findings included otitis media, otitis externa with or without granulation tissue, soft tissue masses and osteolytic lesions of the temporal bone [3].
Contrast enhanced CT proves useful in delineating osseous as well as temporal soft tissue involvement while simultaneously demonstrating destruction of petrous apex. MRI further serves to support the aforementioned information while further clarifying the extent of soft tissue involvement. Confirmation of diagnosis is by means of a tissue biopsy, with immunohistochemistry demonstrating Langerhans’ cells in association with inflammatory infiltrate consisting of lymphocytes, disproportionate eosinophils, giant cells and plasma cells.
Our rare case of extensive multifocal skull base disease illustrates the rapid onset, uncommon nature and variation in presentation of LCH. It is more routinely associated in the paediatric population and with systemic involvement in approximately two-thirds of cases [2, 3]. Our report consolidates further on the limited literature available on LCH-related skull vault lesions within adults while emphasising the crucial role of an otolaryngologist in the early evaluation, staging and subsequent diagnosis of LCH. This is particularly crucial given the age at diagnosis, and initial response correlates closely to prognosis and recurrence. Persistent ear symptoms and atypical localised deep rooted and superficial vault pain should prompt otolaryngologists to incorporate LCH when formulating differential diagnoses. Long-term follow-up in such patients is critical, with relapses likely to occur years beyond initial treatment [5], which may incorporate a conservative medical (chemotherapy, local steroid therapy or bisphosphonates) or a surgical (curette or excision) approach. Radiotherapy is generally reserved for cases of neurological deficits.
Conflict of interest statement
None declared.

{kind=link}