





Abstract
A 61-year-old female with a past medical history significant for von Hippel–Lindau (VHL) syndrome presented with multiple bilateral pulmonary lesions found on surveillance computed tomography scan. Positron emission tomography demonstrated avidity in a lesion in the right upper lobe. After an equivocal biopsy, a lobectomy via a thoracoscopic approach was performed as this lesion was concerning for a primary lung cancer. Pathology revealed a diagnosis of pulmonary mucosa-associated lymphoid tissue (MALT) lymphoma. To our knowledge, this is the first reported case of a pulmonary MALT lymphoma in a patient with VHL.
INTRODUCTION
Extranodal lymphoma of mucosa-associated lymphoid tissue (MALT) can occur throughout the body. While the gastrointestinal tract is the most common site of involvement, about 15% of all MALT lymphomas arise from the lungs [1]. Research over the past several decades has shown that cancers such as lymphomas are a genetic disease and that particular genetic mutations can lead to a syndrome of different cancer histologies. An example of one such syndrome is von Hippel–Lindau (VHL) disease. It is caused by the inactivation of the VHL tumor suppressor gene [2]. While many organs including the pancreas, the kidneys and the central nervous system can be involved in this cancer syndrome, no known association has been established between VHL and primary pulmonary cancers. Here, we describe the presentation and management of a woman with pulmonary MALT lymphoma with a past medical history significant for VHL disease.
CASE REPORT
An asymptomatic 61-year-old woman with no history of tobacco use presented to thoracic clinic with bilateral pulmonary ground glass opacities. She had a past medical history significant for VHL disease (type 2B exon III mutation). Manifestations of her VHL syndrome included a pancreatic neuroendocrine tumor, bilateral pheochromocytomas, spinal hemangioblastomas and benign renal cystic lesions. Upon surveillance computed tomography (CT) imaging of the chest, bilateral ground glass opacities were appreciated. Over a 1-year period, a lesion in the right upper lobe became more solid-appearing (Fig. 1A). Positron emission tomography (PET) imaging revealed that the lesion was also PET-avid with a maximum standardized uptake value (SUV) of 7.7 (Fig. 1B).
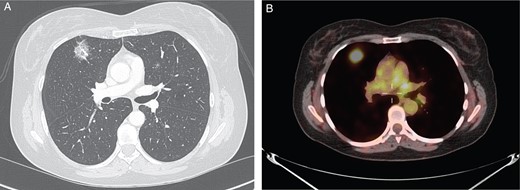
(A) CT chest revealing right upper lobe solid lesion. (B) PET scan of the chest revealing PET-avid right upper lobe lesion.
A percutaneous biopsy was performed, but the cytology report was equivocal, demonstrating rare atypical cells in a background of blood and histiocytes. As this lesion was concerning for a primary lung cancer, a video-assisted thoracoscopic (VATS) right upper lobectomy was performed in an effort to achieve a complete oncologic resection. The operation was carried out in the usual manner, including isolating and dividing the superior pulmonary vein, artery and bronchus with an endo-gastrointestinal anastomosis stapling device. Furthermore, a lymph node dissection, including specimens from stations 10R and 4R, was performed. The procedure was completed without complication, and surgical specimens were sent to pathology.
Grossly, the lymph nodes appeared normal. The largest of the sample was 1.5 cm in the greatest dimension. The microscopic pathology revealed reactive lymph nodes (Fig. 2). The right upper lobe was 194 g and measured 12.5 × 11 × 3 cm. Within it, a 3 × 2.5 × 2.5 cm yellow–tan mass consistent with the area of concern on imaging contained necrotic and hemorrhagic nodules. Microscopically, germinal centers and reactive B follicles were noted (Fig. 3). This was initially diagnosed as a reactive process; but because of the density of the lymphocytic infiltrate and the presence of a mass, molecular studies were performed. These tests revealed a clonal B-cell process consistent with a diagnosis of extranodal marginal zone lymphoma of the MALT type.
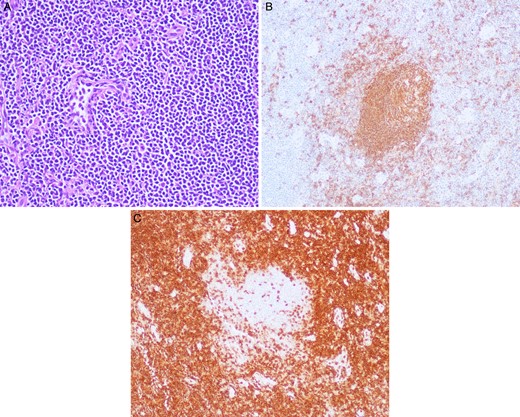
(A) Station 10R lymph node with hematoxylin and eosin (H&E) stain under 20× magnification with many lymphocytes apparent. (B) Station 10R lymph node with CD-20 staining revealing B cells within a reactive germinal center under 20× magnification. (C) Station 10R lymph node with CD-3 staining demonstrating T cells surrounding a reactive germinal center under 20× magnification.
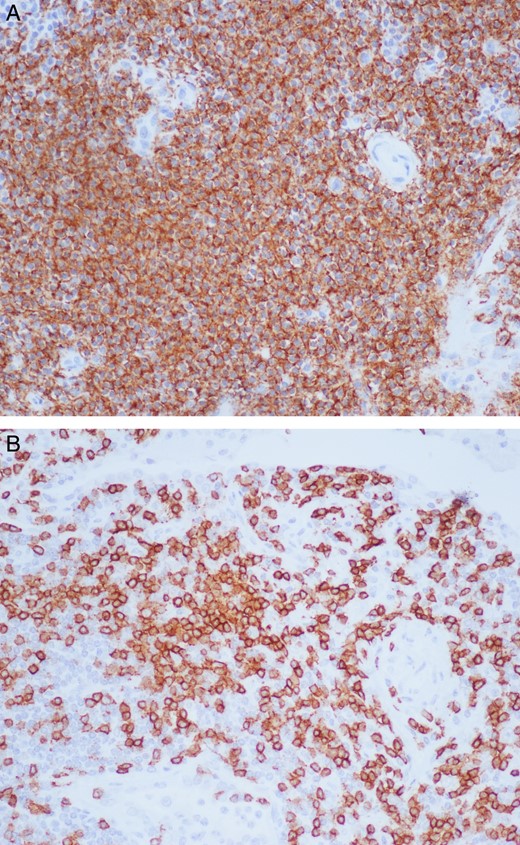
Right upper lobe lung mass under 20× magnification with staining for CD-20 and CD-3 showing a dense lymphoplasmacytic infiltrate mainly consisting of B cells (A) and fewer T cells (B).
The patient recovered well from the operation. She remained in the hospital for 9 days following her surgery secondary to a prolonged air leak, which subsided without intervention. Postoperatively, she will be monitored by both the thoracic surgery and thoracic oncology services for recurrence.
DISCUSSION
Cancer genetic syndromes occur when a single genetic mutation leads to a spectrum of cancer histologies in the same patient. VHL disease is an example of one such syndrome that is inherited in an autosomal dominant fashion and affects 1 per 35 000 people [2]. Inactivation of the VHL tumor suppressor gene via genetic mutations can lead to the development of several different malignant and benign tumors including renal cell carcinoma, pancreatic neuroendocrine tumors, pheochromocytoma, retinal angiomas and spinal hemangioblastomas [2].
Primary lung lymphoma (PLL) is defined as biopsy-proven lymphoma in the lung without the evidence of extra-pulmonary involvement [3] and accounts for <1% of all lung tumors [4]. The most common of the PLLs is MALT lymphoma, which is a low-grade B-cell lymphoma [4]. The diagnosis of pulmonary MALT lymphoma can be mistaken for a reactive process, so additional studies such as molecular analyses to identify clonal populations of lymphocytes may be necessary to confirm the diagnosis [4]. Because of the rarity of the disease, there are no definitive treatment guidelines, but surgery has been shown to be beneficial in patients with localized disease [5].
There is no known association between pulmonary MALT lymphoma and VHL disease to date, but case reports have documented patients with VHL disease who developed other primary lung cancers including adenocarcinomas [6] and carcinoids [7]. Furthermore, loss of heterozygosity of the VHL gene region has been reported in up to 83% of non-small cell lung cancer specimens [8]. However, given the limited number of reports, it is difficult to find a definitive association between VHL disease and primary lung cancers, including pulmonary MALT lymphoma. To our knowledge, this is the first reported case of a patient with VHL disease who developed a pulmonary MALT lymphoma. Whereas the initial pathology report suggested a reactive process, further analysis was consistent with this tumor pathology. Surgeons and physicians in general need to have a high index of suspicion when managing patients with VHL disease and other cancer genetic syndromes in general. Furthermore, increased awareness of these tumor types in patients with familial syndromes may help guide surgical intervention in the future, as a lobectomy may not be necessary, and a non-anatomic wedge resection may be adequate.
CONFLICT OF INTEREST STATEMENT
None declared.

{kind=link}
{kind=link}
{kind=link}