





Abstract
We report the case of a forty-nine year-old Native American female who presented with two-month history of headaches and unsteady gait. MRI brain showed a 6.5 cm by 5 cm enhancing dural-based mass with leftward midline shift. She was taken to the operating room for gross-total tumor resection. Pathology was supratentorial haemangioblastoma. This is the first known case of a supratentorial dural-based haemangioblastoma in a person of Native American descent. We review the literature and describe the unique characteristics of this tumor.
INTRODUCTION
The authors present the eighth documented case of supratentorial dural-based haemangioblastoma in a patient without Von Hippel Lindau Syndrome and the first such case affecting a person of Native American descent. This tumor subtype should be on the differential diagnosis of dural-based tumors in persons of Native American descent without history of von Hippel Lindau Syndrome.
CASE REPORT
Our patient is a forty-nine year-old Native American female with a two-month history of progressively worsening headaches along with gait instability. She reported falling to the left side quite often. She presented to an outside hospital where CT scan of the head showed a large right frontal hypodense mass with significant vasogenic edema and twenty millimeters of left to right midline shift. She was transferred to University of New Mexico Medical Center where MRI of the brain demonstrated a 6.5 cm x 5 cm heterogeneously enhancing cystic and solid mass with significant vasogenic edema and midline shift (Fig 1). Her haematocrit was within normal limits at 40. Family history was negative for von Hippel Lindau Syndrome. VHL Genetic workup was negative and CT abdomen was negative for other lesions.
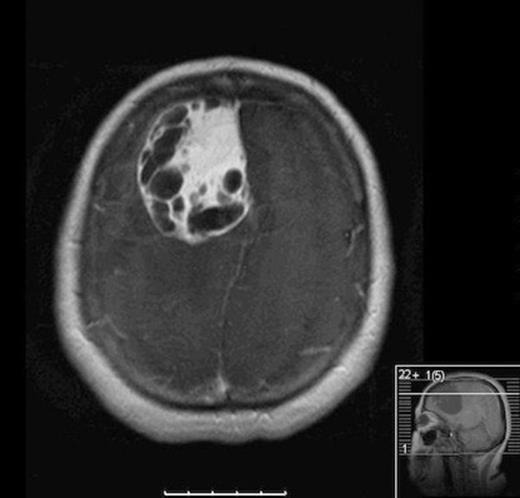
Axial MRI with contrast showing heterogenous enhancing solid and cystic component of tumor
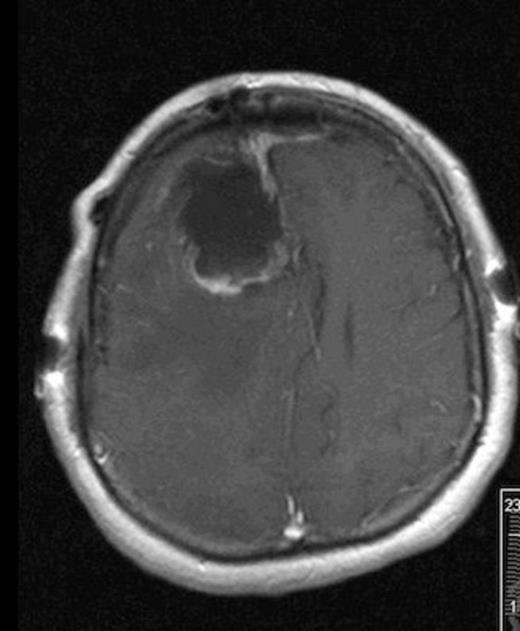
Post-operative MRI showing gross-total resection with some residual blood in cavity
She was taken to the operating the following morning for a bicoronal craniotomy for a gross-total tumor resection with neuronavigation (Fig 2). Pathology was a dural-based WHO Grade 1 supratentorial haemangioblastoma. Microscopic findings demonstrated large vessels and abundant capillary networks, with a cellular proliferation of large stromal cells with clear vacuolated cytoplasm (Fig 3,4,5). There were significant hyperchromatic nuclei and multiple areas of PAS positive pinkish globules (Fig 3,4,5).
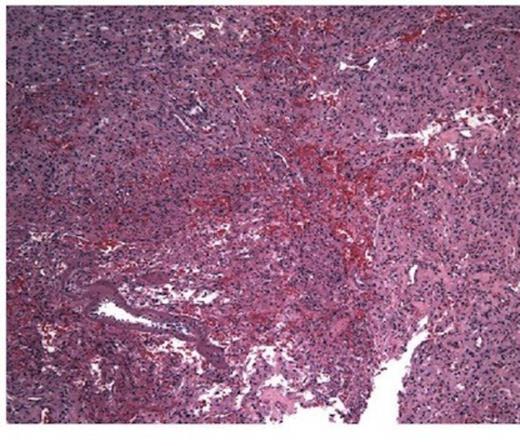
10 x Haematoxylin and Eosin stain showing typical microscopic pathology of capillaries and interstitium
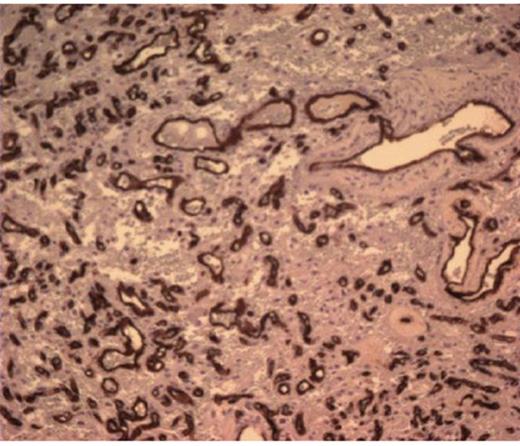
CD 34 positive staining of vascular lining consistent with haemangioblastoma
No mitoses were identified. No whorls or psammoma bodies were identified. Immunohistochemical findings showed interstitial tumor cells immunoreactive for NSE, factor X111A, and S100. RCC, EMA, Inhibin, GFAP, CAM 5.2, CD 10, and CD 34 were negative on the interstitial cells. CD 34 was positive on the vascular endothelium (Fig 3,4,5). She was discharged home within six post-operative days. She returned to clinic one week later and had no neurological deficits.
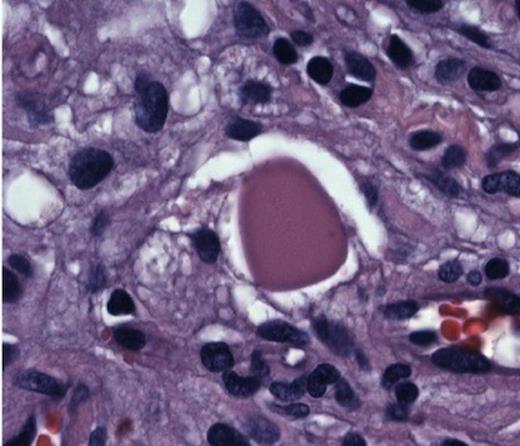
Hyperchromatic nuclei and typical Periodic Acid Schiff + globules at 100x
DISCUSSION
Haemangioblastomas are benign tumors of vascular origin. They constitute about 2 % of all intracranial tumors(2). They are composed of stromal cells of unknown histogenesis and many capillaries (9). In 1938 Cushing, Bailey, and Eisenhardt characterized 23 different vascular meningeal-based tumors into three categories: 1) angioblastic, 2) transitional angioblastic, 3) angioblastomatous meningioma. These were later revised in the 1990’s by neuropathologists Russel and Rubinstein, classifying the variant 3 into haemangioblastoma (3). Lindau had noted in 1927 of the “pseudo-xanthomatous cells” in these tumors and later grown in tissue cultures by Brasseur (4). These likely correlate with the current PAS + globules seen in these tumors.
The association with Von Hippel Lindau Syndrome demonstrates multiple haemangioblastomas, usually infratentorially, and has an autosomal dominant genetic transmission on chromosome 3 (8). These patients usually have family members with a history of intracranial haemangioblastomas, pheochromocytoma, renal cell carcinoma, ocular abnormalities, and abdominal cysts. Most common locations with Von Hippel Lindau Syndrome are in the cerebellum and spinal cord (8).
Supratentorial haemangioblastomas are rare with roughly 116 known cases (3). Supratentorial dural-based haemangioblastomas are exceedingly rare, with only seven published cases in patients without Von Hippel Lindau Syndrome. Note all were VHL negative based on either genetic testing or abdominal imaging. Based on the few cases published, the incidence of males and females are equal with age ranging from 10-62. This case describes the first report of such a tumor in a person of Native American descent. Locations are mostly frontal and parasagittal. However, they can also be found in the parietal lobe as well. The most common reason for admission is headache. Tumor size on presentation varies from 1.5 cm to 7 cm. Isolated lesions occur in 80 % patients with Von Hippel Lindau Syndrome and in 95% of patients without this syndrome(2).
Modern methods of diagnosis of haemangioblastomas includes CT scan and MRI brain with and without contrast. Historically, angiography was used for diagnostic purposes(4). Elevated haematocrit is commonly seen in these tumors with production of erythropoietin(4). Differential diagnoses must include multiple variants of meningiomas, renal cell carcinoma, and haemangiopericytoma(4,5,8,9). Mainstay of treatment includes craniotomy(8). Several published articles describe the effectiveness of gamma knife therapy on treating these tumors long-term(6). Favorable conditions for successful gamma knife therapy included mostly solid tumor without cystic component(6). Even at low marginal dose of 8-30 grey, solid tumors were successfully controlled at up to 146 months follow up(6). Pre-operative embolization has also been described and effective in cases where large vessels can be catheterized successfully.
In conclusion, this is a unique case of a supratentorial haemangioblastoma that should be on the differential diagnosis of dural-based tumors in patients of Native American descent who are without Von Hippel Lindau syndrome. Given the cultural uniqueness and genetic diversity of this population, this report is critical to appreciate when practicing in a region with a predominantly Native American subset such as New Mexico.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}