





Abstract
Rhabdomyosarcoma of the middle ear is a rare tumor, even rarer in adults and has a very poor prognosis. We report here an unusual case of rhabdomyosarcoma in middle ear of an adult, mimicking chronic suppurative otitis media and facial nerve palsy.
INTRODUCTION
Rhabdomyosarcoma is the most common soft-tissue malignancy in pediatric age group arising mainly in the head and neck, most commonly in the orbit and the nasopharynx (1). Ear is comparatively a rare site for this neoplasm and accounts for less than 10% of all cases of head and neck (2). It is an unusual occurrence in adult population, even more rare to involve the middle ear and mastoid (3). Rhabdomyosarcoma frequently looks like a polypoid mass on examination, so easily misdiagnosed as aural polyp and therefore, advanced disease with meningeal involvement is common at the time of diagnosis (1).
We report here an unusual case of Rhabdomyosarcoma of middle ear and mastoid in a 31-year-old man.
CASE REPORT
A 31 year old man presented in the ENT outpatient department with complaints of left sided facial asymmetry, inability to close left eye and discharge of fluid from left ear for the past 2 months.
On examination the patient had left sided facial nerve paresis and bloodstained discharge was seen in external auditory canal. Examination under microscope revealed a mass in middle ear, which was bleeding on touch.
High Resolution CT scan of temporal bone revealed a contrast enhancing mass in the left middle ear eroding the ossicles (Fig. 1a). A biopsy was taken from the middle ear.
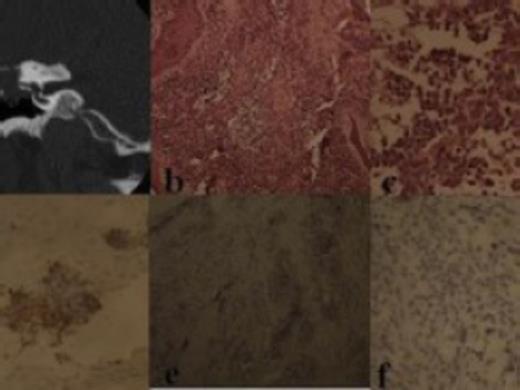
a) CT scan showing mass in the middle ear; b) Photomicrograph showing tumor tissue surrounded by stratified squamous epithelium and admixed with fibrous stroma (H & E, X 100); c) Photomicrograph shows discohesive, round to oval neoplastic cells with scant cytoplasm and hyperchromatic nuclei (H & E, X 400); d) Photomicrograph showing immunopositivity of neoplastic cells for desmin (X 400); e) Photomicrograph shows immunonegativity for CD20 (X 100); f) photomicrograph shows cells also negative for CD3 (X 400)
Histopathological examination of the paraffin embedded sections revealed tumor tissue, surrounded by stratified squamous epithelium (Fig. 1b) and composed of nests of discohesive, round to oval neoplastic cells (Fig. 1c), with scant, eosinophilic cytoplasm and hyperchromatic and pyknotic nuclei present in a loose fibrous stroma, at places firmly attached to fibrous strands. Considering all these features, a diagnosis of a small round cell neoplasm, probably embryonal rhabdomyosarcoma was considered and immunohistochemistry was performed. Immunohistochemically, the neoplastic cells were positive for desmin (Fig. 1d) and negative for CD20 (Fig. 1e) and CD 3 (Fig. 1f), thus confirming the diagnosis of embryonal rhabdomyosarcoma.
The patient was treated with chemoradiation but developed recurrence and expired six months after initial diagnosis.
DISCUSSION
Rhabdomyosarcoma is one of the most common tumors of pediatric age group (1). They exhibit a bimodal pattern of age distribution: peaking between 2 to 5 yrs age group and then a spike in late adolescence (1). Overall 63% cases are under 10 yrs of age (2). Based on histology, it is classified into embryonal, alveolar, pleomorphic, and mixed histologic subtypes (3). Embryonal rhabdomyosarcoma is the most common histologic variant (3).
Middle ear cleft is a rare site for this tumor, more so in adult population (3). To add to this, signs and symptoms of this area are similar to chronic suppurative otitis media, thereby delaying the diagnosis (2).
Clinical features include blood stained purulent ear discharge, impairment of hearing, earache, aural polyp and granulations and, in advanced cases, neurologic symptoms (4). These features being nonspecific, can lead to a delay in diagnosis. As a result, most of these patients are initially managed on antibiotics and only when this treatment fails is some other diagnosis sought for (4).
These patients also progress faster to neurological problems due to erosive nature of disease; most common neurologic problem being facial nerve involvement. In advanced cases it is not uncommon to see meningeal or brain parenchyma involvement (4).
Tissue diagnosis is the important and should be done as soon as there is any doubt regarding diagnosis (5). Routine histopathological examination shows a round cell tumor (5). Immunohistochemistry is required to differentially diagnose it from other small round cell neoplasms, namely lymphoma (CD20, CD3 positive) and Ewings sarcoma/PNET (CD99 positive) as Rhabdomyosarcoma is negative for these markers and diagnostically positive for desmin is diagnostic of rhabdomyosarcoma (5).
The treatment for this neoplasm has been a controversial issue. Before 1972, the mainstay of treatment was surgical excision followed by radiotherapy. Intergroup Rhabdomyosarcoma Study (IRS) protocol was brought out in 1972, which drastically shifted the paradigm to multiagent chemoradiation. It was advised that surgical resection should only be done if major morbidity can be avoided. This protocol was advised in 1978 (IRS II), 1984 (IRS III) and 1991 (IRS IV)(1). The basic difference is the increasing and better role of chemotherapeutic agents. Newer agents, etoposide, ifosfamide and melphalan have been added to already existing standard regimenof vincristine, actinomycin D and cyclophosphamide (6). Radiotherapy delivered to head and neck is usually limited to 4000 to 4500 Gy (6). Salvage therapy for recurrent rhabdomyosarcoma is problematic (3).
Basic goal for treatment is locoregional control and prevention or treatment of distant metastases (3). Systemic treatment with chemotherapy and locoregional control with radiotherapy and if needed surgery is helpful (3). Surgery is not possible in all cases in areas involving vital structures and intracranial extension (1,6).
To conclude, this case is presented to emphasize the unusual occurrence of rhabdomyosarcoma in the middle ear of adults as well, which may mimic chronic otitis media. So a high index of suspicion is required for such cases to prompt early diagnosis and treatment.

{kind=link}