





Abstract
AgBase provides annotation for agricultural gene products using the Gene Ontology (GO) and Plant Ontology, as appropriate. Unlike model organism species, agricultural species have a body of literature that does not just focus on gene function; to improve efficiency, we use text mining to identify literature for curation. The first component of our annotation interface is the gene prioritization interface that ranks gene products for annotation. Biocurators select the top-ranked gene and mark annotation for these genes as ‘in progress’ or ‘completed’; links enable biocurators to move directly to our biocuration interface (BI). Our BI includes all current GO annotation for gene products and is the main interface to add/modify AgBase curation data. The BI also displays Extracting Genic Information from Text (eGIFT) results for each gene product. eGIFT is a web-based, text-mining tool that associates ranked, informative terms (iTerms) and the articles and sentences containing them, with genes. Moreover, iTerms are linked to GO terms, where they match either a GO term name or a synonym. This enables AgBase biocurators to rapidly identify literature for further curation based on possible GO terms. Because most agricultural species do not have standardized literature, eGIFT searches all gene names and synonyms to associate articles with genes. As many of the gene names can be ambiguous, eGIFT applies a disambiguation step to remove matches that do not correspond to this gene, and filtering is applied to remove abstracts that mention a gene in passing. The BI is linked to our Journal Database (JDB) where corresponding journal citations are stored. Just as importantly, biocurators also add to the JDB citations that have no GO annotation. The AgBase BI also supports bulk annotation upload to facilitate our Inferred from electronic annotation of agricultural gene products. All annotations must pass standard GO Consortium quality checking before release in AgBase.
Database URL:http://www.agbase.msstate.edu/
Introduction
Among databases and resources that provide literature-based biocuration, there are two broad approaches for targeting biocuration. In the first approach, biocurators regularly triage all published literature to identify articles that are likely to contain information to be biocurated. This approach works particularly well where the literature is focused to a well-defined set of journals, and there is a larger research community. In the second approach, biocurators target certain gene sets and, for each gene in this set, do comprehensive literature searches to identify all annotation for this gene. Using this approach, databases can target well-studied gene sets and biocurators are able to provide a comprehensive annotation set for a gene or gene product. Naturally, these approaches are not exclusive; as biocurators from different databases collaborate to provide coordinated and consistent annotation, biocurators may change their biocuration approaches to suit their needs.
The AgBase database provides functional data for agricultural researchers through both sequence-based functional annotation and manual biocuration of published literature (1). Currently, our literature annotation is focused on providing Gene Ontology (GO) annotation for agricultural gene products from chicken, cow, corn and cotton and Plant Ontology (PO) (2) annotation for cotton. We utilize GO and PO Consortium best practices and procedures while adapting our biocuration process to the specific needs required for agricultural literature. This body of literature differs from that of model organism species in that it includes a large body of work addressing general production issues, genetic markers and trait characterization; data that does not typically contain functional information. In addition to identifying gene products that have been studied directly in agricultural species, we must also identify which of these gene products are likely to have literature that will yield functional annotations. Moreover, we wish to identify genes that are important for the agricultural research community and provide GO annotation to support further functional modeling of large scale data sets. As a result, we do not attempt to provide detailed, literature-based functional annotation for every gene product but rather focus on annotating prioritized gene sets for the agricultural species on which we work.
With less funding for model organism databases and expanding genomic capacity, the need to rapidly develop simple biocuration interfaces (BIs) for species that do not have the large infrastructure and investment traditionally available to model organism databases is becoming more important. The workflow described here was developed in a modular way, as each of these modules became important to our curation effort. We are not suggesting that other databases would wish to exactly duplicate our workflow but rather they may wish to consider each of these modules in the context of what they add to the curation workflow (e.g. ability to prioritize curation, identify literature and track journal requirements).
Biocuration workflow
This article describes the pipeline we developed to prioritize our annotation effort, ensure that we rapidly and accurately identify literature for GO annotation, capture annotation and literature details, do quality checking and generate gene association files (Figure 1). Our goal in describing our workflow is 2-fold. First, we wish to inform researchers who seek to use AgBase how we prioritize our curation effort and handle their requests so that they are better able to get the annotation that they need to do functional modeling of their own data sets. We wish to make our curation goals transparent to the research community and are working to make the gene prioritization (GP) interface visible to those who request annotation. Moreover, as we develop links from our text-mining tool to our main database, researchers will see functional terms and links to PubMed articles appear on AgBase gene product pages along with functional annotation. An understanding of the Extracting Genic Information from Text (eGIFT) tool and how we use it in AgBase biocuration will enable researchers to better understand the functional information available for gene products, regardless of whether it is annotated to an existing ontology. Second, we wish to demonstrate how text mining can be integrated into a curation workflow to identify literature for manual curation. The eGIFT tool has already been published (3) and is available online and can be incorporated into existing curation interfaces via web links.
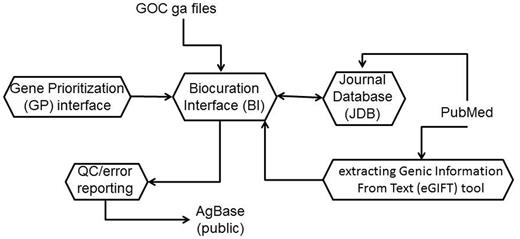
The AgBase biocuration pipeline. The AgBase biocuration pipeline draws from GO Consortium gene association files and from PubMed data, and the output is the AgBase public gene association files. Briefly, genes to be annotated are prioritized as a ranked list in the GP interface, which are linked to records in the main BI. The eGIFT tool enhances the ability for biocurators to identify and curate appropriate literature while the JDB records reviewed literature. Biocuration must pass standard GO Consortium error and quality checks before public release.
The individual components of this pipeline are described in more detail in the following sections. Currently, we are working to integrate PO annotation into the existing AgBase curation flow.
Gene Prioritization interface
As with all databases, AgBase biocurators seek to focus their curation efforts on gene products that will be of most benefit for the research communities they serve. Researchers who had the most need for GO annotation when we started our curation were those who had microarray data to functional analyze. We surveyed agricultural researchers to identify which microarray platforms they used (or were planning to use) and focused on providing annotation for gene products on the array platforms commonly used by our communities. Because many of these arrays had array annotation files that were out of date, we first updated all public database accessions cross-referenced to the array probes to remove or update any obsolete database records. (This updated ID mapping is available at the Array Annotation link on AgBase.) Next, we associated existing GO annotation the gene products represented by the array probes and used these same accessions as our targets for further GO annotation. AgBase users may access the array ID mapping and GO annotation files via the Array annotation link or can access GO annotations for their own data sets (including array data) via the GORetriever tool on AgBase.
An additional consequence of our community survey is that we are approached by researchers who have either experimental data sets or their own arrays which they wish to have GO annotated. We encourage researchers to contact AgBase directly with requests for annotation of specific gene sets to support their own experimental analysis. Naturally, our ability to provide additional GO annotation is dependent on the time for curators to do annotation, the amount of annotation requested and what published literature is available for curation. Over the past 5 years, we received an average of six requests for annotation of data sets with more than 1000 gene products. However, as more agricultural researchers move from arrays to RNASeq expression data, the data sets we assist with are increasing in size (from 2000 to 3000 differentially expressed transcripts to gene sets of 20 000–30 000 in RNASeq data sets) and the diversity of species being studied is also increasing. This increase is offset because many genes in many agricultural species have no published literature and can only be GO annotated based on computational analysis of sequence, which is faster than literature-based curation. Nevertheless, detailed, manual curation of genes that have published data would greatly assist in computational-based propagation of function to genes identified by new sequencing technologies. So that researchers can understand our curation goals; our process for prioritizing GO annotation is described below.
AgBase biocurators target manual biocuration using a GP interface (Figure 2) that ranks genes based on user requests or presence on microarrays. Gene information is loaded from the NCBI Entrez Gene database, and a prioritized gene list is generated for each species (Figure 2A) for which we provide literature annotation. In each case, genes are ranked using a simple scoring system where they are assigned a count for each time they have a gene product that occurs on commonly used microarrays. When researchers request annotations via the AgBase Community Requests and Submissions page, the count is also incremented. Researchers who request annotations are provided with a view only link to access the relevant GP list so that they can track their request in the queue. Prioritization based on a mixture of functional genomics platforms and researcher requests enables GO annotation for each species to reflect community needs and current interests. In addition, biocurators can also add annotation requests (e.g. as part of a collaborative project to annotate defined gene sets).
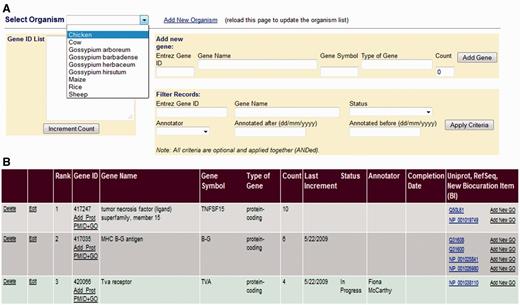
The GP Interface. The GP Interface is used to direct biocurator’s annotation to genes that the community see as requiring annotation. Genes are prioritized separately for each species (A), and each species has a searchable, ranked list where genes are ranked based on requests for annotation and presence on commonly used array. Each gene is linked its gene products in the biocuration interface (B) so that the biocurator can move seamlessly to annotation.
Biocurators with access to the AgBase biocuration pipeline access the GP interface and select the species on which they are working. The GP interface shows the ranked list of genes, gene names, their current count and links to the corresponding gene product in the main BI (Figure 2B). To ensure that each biocurator can track not only their own annotation but also ongoing biocuration by others in the group, a biocuration status menu is displayed. The status menu can be set to display ‘annotation in progress’ or ‘annotation complete,’ and the name of the biocurators (based on login) is displayed. The biocurator selects the top-ranked genes, sets the menu to indicate that they are working on this gene and uses the gene product link to go directly to the corresponding gene product in the main BI. Once the gene is marked as complete, its count is zeroed and the date recorded.
Each GP list is updated as part of the regular AgBase database update procedure. Genes with a count that become obsolete are flagged for biocurators’ review and reassignment, if possible. This process helps us ensure that the AgBase mappings from array IDs to gene product accessions remain current.
Biocuration Interface
The main BI may be accessed via the GP interface or directly, the latter method enabling biocurators to also add GO annotations for gene products that do not have a specific GP list. Agricultural literature that we curate often contains functional data for more than one gene product or species and it is our policy to annotate all GO functional information from papers that we manually curate.
Our BI is specifically designed for GO annotation and is updated to ensure that we are able to capture additional data fields mandated in the new GO gene association file format (gaf 2.0). With each update (done every 2 months), we load all manual GO annotations provided by the GO Consortium and computationally derived GO annotations [Inferred from Electronic Annotation (IEA)] for those species that we specifically target and updated GO term information. By loading species-specific IEA annotations provided by European Bioinformatics Institute Gene Ontology Annotation (EBI GOA) biocurators are able to see at a glance the likely GO terms that they may expect to find in their literature searches. By loading manual annotations for other species, we are able to transfer GO annotations among species where there is evidence of sequence or structural similarity.
We use standard quality checking scripts developed by the GO Consortium to prompt biocurators during the annotation process; biocurators are prompted to ensure that all mandatory fields are completed, they are completed in the expected format, GO IDs are not obsolete and so forth. Information about the biocurator and the date of annotation are auto filled. Because we are annotating species for which there are no model organism databases or reference gene set and we do not restrict our annotation to proteins from UniProtKB, biocurators may also add gene product record pages using accessions from NCBI if there is no UniProtKB record available or in cases where the protein record is not appropriate (e.g. functional RNAs). Another feature that we found necessary for our biocuration pipeline was the ability to add literature records not found in PubMed. However, PubMed does not contain all agricultural functional literature, so we allow biocurators to add references from agricultural literature collections such as Agricola, the National Agricultural Library Catalog. GO Consortium guidelines allow this practice, although where possible we prefer to use PubMed IDs. We also include a free text comment field for biocurators to capture additional, pertinent information. Most typically this is used to note a new GO term request, or until the recent change in the GO gene association file, to capture links to other ontology terms. AgBase biocurators request new GO terms as required using the GO Consortium SourceForge new term request.
eGIFT integration
Another novel tool that we use to focus our manual biocuration effort is the eGIFT tool (3). eGIFT is a web-based, text-mining tool that associates informative terms (iTerms) with genes based on text from PubMed abstracts. Briefly, the eGIFT system retrieves a set of abstracts associated with a gene and identifies iTerms by scoring single- and two-word terms for the abstract set as well as GO term names, NCBI taxonomy, Unified Medical Language System (4) and Medical Subject Heading terms. Each iTerm is given a score that reflects that iTerm’s relevance to the gene’s set of abstract compared to the set of background abstracts. Where possible, iTerms are categorized based on the different ontologies and vocabularies used to retrieve the abstract set (e.g. GO molecular function/biological process, GO cellular component, domains and motifs, diseases), and results are presented as iTerms grouped by categories in descending order of score for each gene or as a gene list for each iTerm. Because most agricultural species do not have standardized nomenclature and gene names can be ambiguous, identifying literature associated with a particular gene can be problematic and time consuming. Note that eGIFT developers chose to focus on PubMed abstracts because abstracts are a better source of keywords than full-length articles (5), and our purpose here is to identify relevant PubMed articles, rather than use text mining to identify ontological terms for curation.
We incorporate eGIFT into our annotation pipeline by displaying this information in the top right hand corner for each gene product page of our BI (Figure 3). As a biocurator moves from the GP to its linked gene product page in the BI, they immediately see a summary of functional literature associated with the gene. As each eGIFT iTerm is linked to corresponding GO terms, the eGIFT information is easily displayed as a list of GO terms found in the literature for that gene with links to the associated literature and additional links to the full eGIFT information page. Thus, the BI page displays not only existing GO annotation but also possible functional literature classified by GO terms. This enables the biocurator to rapidly assess which eGIFT-predicted GO terms may already be annotated to the record and what additional functional information they may capture. Displaying this information on one page means that the biocurator can rapidly move from gene selection to annotation of literature, without intervening literature triage or searching.
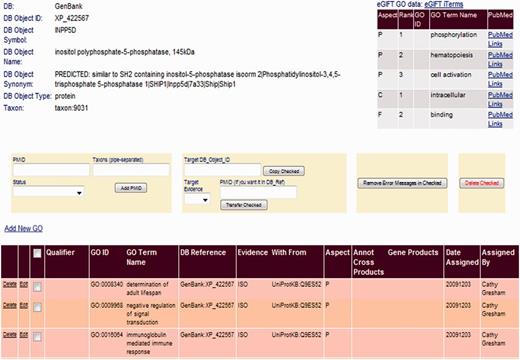
Linking eGIFT to the AgBase BI. A summary of eGIFT GO terms and links to corresponding literature is displayed in the top right hand corner of each gene product page in the AgBase BI. This table allows biocurators to rapidly identify potential new GO Terms and link out to relevant literature.
Journal Database
Like many other biocuration projects, we also track the literature that we annotate. Because we initially had difficulty in identifying literature that contained GO annotation and literature is only poorly associated with agricultural genes in any case, we sought to collect information about the articles that we identified for annotation. We developed the AgBase Journal Database (JDB), which we have subsequently integrated into our biocuration pipeline. Although we intended this latter feature to measure our ability to identify functional literature, it also provides a source of manually curated true negatives for text-mining projects.
When a biocurator enters an annotation in the BI that contains a PubMed reference, these data are also recorded in the JDB. The JDB collects the PubMed ID, authors, title, journal and citation details and links out to the PubMed record. However, we also record articles that have no GO annotations. Specifically, these are articles that we initially identified for GO annotation based on inspection of the abstract text that, on reading the full-length article, were found to have no GO annotation information. This ability to record articles that do not have GO is a key feature of JDB and it differentiates it from other JDBs for biocuration projects of which we are aware. Again, because there are no model organism databases for the species that we annotate and no dedicated effort to link literature to genes and gene products, AgBase biocurators also routinely submit NCBI Gene Reference Into Function notes, most particularly when the literature they identify is not already linked to the Entrez Gene record.
The third type of data (other than articles with and without GO) that we capture in the JDB is articles that we identified as likely to contain GO but which we were unable to access via our current library subscription; we send this information to our institutional library so that they are able to receive feedback about journal subscriptions we require. The JDB not only provides links between genes products and literature but also used to analyze which journals we frequently use (or would wish to use if access were provided).
We also provide a JDB free text comment field that can, for example, be used to note information that may be relevant to ontologies other than the GO (most commonly tissue expression, cell type and post-translation modifications).
Currently, there are (as of July 2012) 712 articles in the JDB, and 80.8% of these included GO annotation information while 17.8% had no GO annotation (Figure 4). The overall number of articles that we add to JDB varies from year to year based on the number of biocurators, and the proportion of articles that are marked as having no GO annotation is a reflection of the ability of these biocurators to accurately identify articles that will contain GO data. For example, we first received funding to hire biocurators in late 2007, and the number of articles submitted to JDB increases dramatically after this time. Also, the largest proportion of articles marked as ‘no GO’ are found during 2008 when biocurators were learning to identify articles for annotation. In 2011, we implemented eGIFT to assist biocurators with identifying articles for annotation.
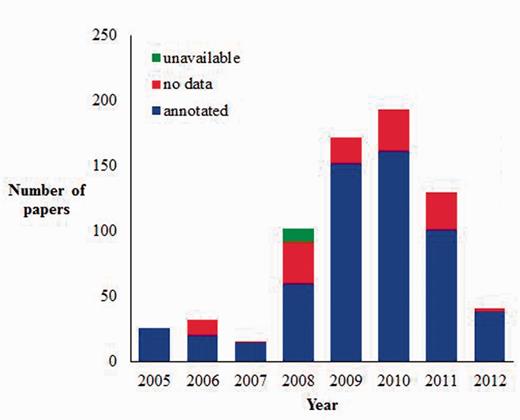
Current AgBase JDB statistics (as of July 2012). AgBase biocurators record the articles they look at for biocuration in JDB and classify them as annotated (contain information they annotate to the GO), no data (contain no GO data) or unavailable (likely to have GO data but unable to obtain full article for curation).
Bulk annotation upload
In addition to providing an interface that allows biocurators to add annotations directly, we also provide a bulk annotation upload feature that enables us to add our sequence-based annotations from computational annotation pipelines, such as those generated by InterProScan analysis and mapping to GO. During the upload process, annotations are quality checked to ensure that they meet current GO Consortium standards and that they are not duplicating existing GO annotations. Biocurators have the option of selecting either to discard duplicated annotations from the upload file or to use these annotations to override existing annotations in the BI with the newer annotations. Any annotation errors are flagged for manual review before loading into the BI.
Quality checking and error reporting
As mentioned earlier, our biocuration workflow does quality checking and error reporting at two stages—during the initial annotation entry (either via the BI or as a part of the bulk upload process) and again during the regular AgBase updates. As part of the regular AgBase updates (done every 2 months), new annotations from other GO Consortium members are loaded into the BI and the GO files (including GO:IDs, term names, etc) are reloaded so that curators have access to updated GO term information for their curation.
During the AgBase update process, the quality checks are more extensive and only annotations that pass are forwarded to the public AgBase database. Annotations that are flagged as errors are passed to the associated Error Reporting interface where biocurators may view these based on either the type of error or by biocurator; this enables biocurators to manually review and correct any annotation error before their public release. The types of errors that we check include errors in the GO Consortium–produced GO annotation file checking script, as well as internal checks to ensure that our annotations map to existing records in AgBase and are not duplicated within our workflow. Examples of errors include mandatory annotation fields not completed; unrecognized field formats; use of obsolete GO IDs; instances where an external accession is incorrectly linked to more than one record in the BI; and automatic checks to ensure that annotations transferred based on sequence or structure homology are still relevant.
GO annotations that pass our standard quality checks and the quality checks using the GO Consortium–produced GO annotation file checking script are made public on the AgBase website. Our manual GO annotation is also entered in the EBI GOA Protein2GO system, where it is collected by the GO Consortium. However, not all our GO annotations can be passed through the EBI curation interface; this interface is limited to proteins, and initially to proteins that had a UniProtKB accession, while we also annotated some NCBI gene products with no equivalent UniProtKB accession. Moreover, this interface did not handle newer GO evidence codes. During the period when EBI was working to add functionality to its annotation interface, we used this interface to submit manual annotations that it supported and made the remainder of our annotations public via our ‘AgBase Community’ gene association file. Improvements to the EBI GOA Protein2GO interface and an initiative by the GO Consortium to develop accessible GO curation interfaces are expected to circumvent this problem in the future and encourage dissemination of manual GO annotations obtained via new sources.
Generating annotation reports
Another feature linked to our biocuration workflow is the ability for biocurators to quickly generate annotation reports and subsets of annotations contained in our BI. We do this by allowing them to set up Boolean searches based on the gene association file fields (including date and submitter). This allows biocurators to rapidly report how many GO annotations exist for a particular species, how many GO annotations were added during a specific time frame or to quickly find specified sets of annotations that they added. This simple interface reports both the number of GO annotations and the gene product records returned by a search, as well as allowing the results to be downloaded as a gene association file.
Implementation
The software supporting the AgBase workflow is implemented as a web-based, password-protected Intranet. The system is implemented in ASP.NET with a MySQL backend. It incorporates three local databases (GP database, BI database and JDB) and also has read access to the remote eGIFT database via web services. These three databases provide and capture the information necessary for the individual components described earlier (Figure 5). The GP database is populated by loading the data from the gene_info files from Index of ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/GENE_INFO/ for the desired species AgBase biocurators are annotating. Also, a table with user-specified gene to protein mapping is maintained in the GP database. Because GO annotation is done for all proteins associated with a given gene, the gene to protein mapping is added by loading data from gene2refseq and gene_refseq_uniprotkb_collab files from ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/. The BI database contains two GO gene association datasets. The first dataset contains our AgBase annotations. We also load GOA Uniprot data from http://www.ebi.ac.uk/GOA/uniprot_release.html so that biocurators can see existing GO annotations provided by other GO Consortium groups. The publication data for the JDB is either fetched from NCBI based on the supplied PubMed IDs using NCBI E-utilities (http://www.ncbi.nlm.nih.gov/books/NBK25500/) or entered manually if an article does not have a PubMed ID. The eGIFT DB is located remotely and incorporated into the AgBase biocuration workflow via web services. These databases are updated every 2 months as part of the standard AgBase update and quality check procedures.
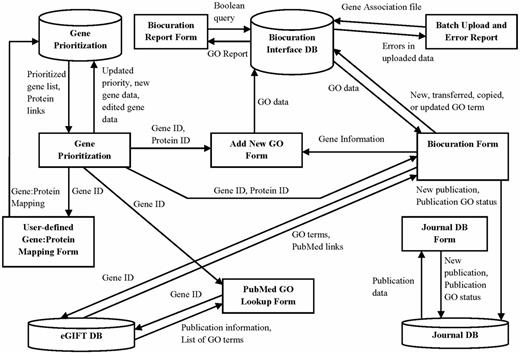
The AgBase annotation workflow is supported by three underlying databases. This schema shows the GP, BI and JDB (cylinders), the information they contribute to interface forms (squares) and the data from each database used to create these interfaces (arrows).
Future work
Our future work focuses on extending the AgBase BI to include annotation to multiple ontologies. Currently, we are working on extending our interfaces to include PO annotation data. In addition, by analyzing the eGIFT iTerms that do not map to GO terms, we are able to identify information in the literature that we are not currently capturing. (For example, preliminary analysis of chicken-based iTerms identifies information about tissue expression and anatomical development.) Future AgBase biocuration will require interface that are extensible to other ontologies. We are also developing methodology to use eGIFT to inform our GP interface by analyzing which genes have the most functional literature available. Moreover, to ensure that biocurators’ time is used effectively and efficiently, we will require improved visualization and human–computer interaction analysis of these interfaces.
Funding
This work was supported by the US Department of Agriculture (USDA) Agriculture and Food Research Initiative (2011-67015-30332), Cooperative State Research, Education and Extension Service (2007-35205-17941 and 2008-35205-18734) and the National Institutes of Health National Institute of General Medical Sciences (07111084). Functional annotation of cotton is supported by USDA Agricultural Research Service (58-6402-7-241). Funding for open access charge: National Institutes of Health National Institute of General Medical Sciences (07111084).
Conflict of interest. None declared.
References
Author notes
†These authors contributed equally to this work.
Present address: Fiona McCarthy, Department of Veterinary Science and Microbiology, University of Arizona, Tucson, AZ.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}