





Abstract
FlyBase is the model organism database for Drosophila genetic and genomic information. Over the last 20 years, FlyBase has had to adapt and change to keep abreast of advances in biology and database design. We are continually looking for ways to improve curation efficiency and efficacy. Genetic literature curation focuses on the extraction of genetic entities (e.g. genes, mutant alleles, transgenic constructs) and their associated phenotypes and Gene Ontology terms from the published literature. Over 2000 Drosophila research articles are now published every year. These articles are becoming ever more data-rich and there is a growing need for text mining to shoulder some of the burden of paper triage and data extraction. In this article, we describe our curation workflow, along with some of the problems and bottlenecks therein, and highlight the opportunities for text mining. We do so in the hope of encouraging the BioCreative community to help us to develop effective methods to mine this torrent of information.
Database URL:http://flybase.org
Introduction
FlyBase is the leading database for genomic and genetic information on the model organism Drosophila melanogaster and other members of its clade (1). The website contains over 2.5 million pages covering 19 different data classes and 20 Drosophila-sequenced genomes. FlyBase displays data on over 144 000 alleles, themselves associated with over 30 000 gene records. Allele and gene reports accommodate phenotypic, genetic interaction and molecular data, much of which are extracted from the published literature by highly trained curators. FlyBase curators capture information from the published literature and integrate this with genome annotation, high-throughput datasets, stock information and other bulk datasets to provide a one-stop site for Drosophila-related information.
Research trends and methods are constantly changing, and FlyBase curation has evolved over the last 20 years to accommodate these changes. During this period, the number of Drosophila-related primary research articles published each year has steadily increased, from roughly 1000 in 1980, to over 2000 a year since 2001 (2). FlyBase genetic literature curation is performed on an article-by-article basis (as opposed to gene-by-gene or topic-by-topic) and until 2008, we used a variety of tier systems to list and prioritize journals for curation. The advantage of curating in an article-by-article manner is that each article is seen by one curator, rather than by a triaging curator and then a number of different curators, each curating a different aspect of the article (e.g. one for genetic interactions, one for phenotypes). This is more efficient and can lead to improved accuracy in curation. For example, Gene Ontology (GO) annotations are often taken from many parts of an article and can sometimes be formed only by taking into account information from different sections and figures. Between 2005 and 2008, we had a three-tier system whereby journals (and the articles therein) were ranked based on a combination of their impact factor and an experienced-based estimation of the quantity of FlyBase-curatable Drosophila-related information they contained. We had a watch list of ∼35 journals and each curator would select a journal from the list and inspect its latest issue in order to identify relevant articles. This method was not perfect however, as many data-rich articles from non-priority journals were only curated when we were alerted to them by users or from citations in other articles.
Novel strategies were needed to identify relevant data from a wider spectrum of Drosophila literature and to rank articles by criteria more specific than parent journal. In 2008, we introduced our first-pass ‘skim’ curation triaging system, where we extract a minimal level of information from every article and reserve full genetic curation for the more challenging data types. We brought authors into the process in 2011 with the introduction of an ‘email-author’ system, whereby we contact authors of recently published articles and ask them to fill in an online ‘Fast-Track Your Paper’ (FTYP) form that recapitulates our skim curation (2). In the first 9 months of e-mailing, corresponding authors skim curated 44% of newly published articles. We then began sending reminders to authors who have not responded and author-led skim curation has increased to 57%.
Another improvement we are exploring is the addition of text mining to the FlyBase curation pipeline. The continued increase in article number, data types and data density (within each article) is an on-going challenge to curation management. If software can shoulder some of the burden of paper triage and data extraction we can devote more curation time to the specialized task of full curation. This overview of FlyBase genetic literature curation practices highlights promising targets for effective text mining of Drosophila data.
The genetic literature curation workflow
It can take between 2 and 4 months on average for an article to be fully curated, from deposition in NCBI’s PubMed to being available on our public web server (Figure 1, curation pipeline). We perform a semi-automated search of NCBI-PubMed for Drosophila-related publications every week. The title and abstract for each publication are checked by eye to confirm that the publication contains Drosophila-related data, after which we semi-automatically add the basic citation details to the FlyBase database. The corresponding authors of these recently published articles are then sent an email inviting them to use our FTYP form to skim curate their article. After a suitable delay to allow the authors to respond, we then generate a list of all the publications that haven’t yet been either fully or skim curated. We do this by not only using those citations that we have just added, but also those citations that we haven’t yet curated from previous citation loads. This means that this list contains all the ‘untouched’ articles, i.e. all those that haven’t been curated in any form, either by us or by authors through our FTYP tool. Each article on this list undergoes a first-pass ‘skim’ curation by FlyBase curators. As the name suggests, skim curation is a shallow, quick curation, where the main genes mentioned within an article are associated with that publication. We also flag the article for the data types present, such as whether the authors generate a new allele or whether there is expression data in the article (we call this second step ‘triaging’) (Table 1). These first-pass curation records are loaded into the database and made available to the public in the next release of FlyBase so that users can find all the references associated with their gene of interest.
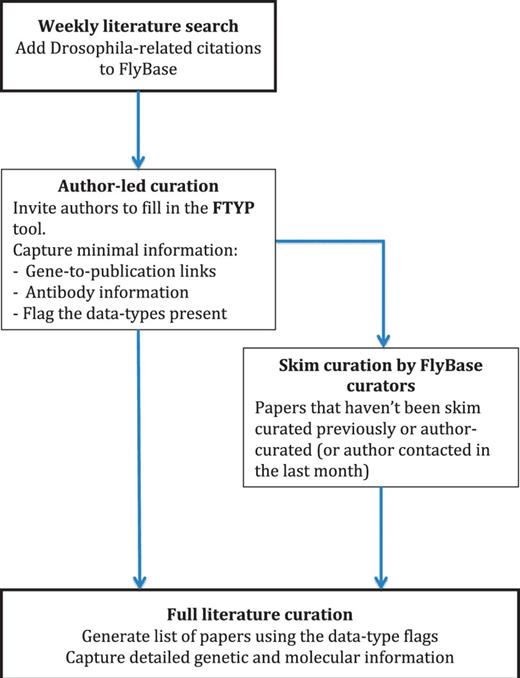
Literature curation pipeline. A weekly PubMed database search identifies new Drosophila-related publications. The citation details for these articles are added to our bibliography, and where possible, the associated PDF is downloaded. The correspondence email for each new article is extracted from the PDF and used to invite the author to use our FTYP tool. Through use of this tool, or a FlyBase curator ‘skim’ curating each article, gene-to-publication links are generated. These are published to our FlyBase website at the first opportunity. Data types found in the article, flagged either by the authors or curators, are then used to generate a priority list for ‘full curation’, where we extract detailed genetic and molecular information.
Data-type flags used in literature curation (taken from an article by Bunt et al., 2012; see Ref. 2)
| Data-type flags | Data presented in an article |
|---|---|
| Drosophila reagents | |
| New allele or aberration | Generation of a new classical allele or chromosomal aberration in a Drosophilid genome |
| New transgene | Generation of a new transgenic construct |
| Gene characterization | |
| Initial characterization | Initial characterization of a Drosophilid gene |
| Merge of gene reports | Evidence suggesting the merge of two or more FlyBase gene reports |
| Gene rename | New gene symbol or name for an existing gene in FlyBase |
| Expression | |
| Expression in a wild-type background | New temporal or spatial expression data of any D. melanogaster gene in a wild-type background |
| Expression in a mutant background | Expression data of any D. melanogaster gene in a mutant background, or after environmental perturbation |
| Phenotypes and interactions | |
| Phenotypic analysis | Novel phenotypic data |
| Physical interaction | Physical interactions involving D. melanogaster proteins or nucleic acids |
| Genome annotation data | |
| Changes to D. melanogaster gene model | New experimental data relevant to D. melanogaster gene model structure |
| Changes to non-D. melanogaster gene model | New experimental data relevant to the gene model structure of non-D. melanogaster Drosophilid genes |
| Mapping of features to genome | D. melanogaster molecular mapping data |
| Cis-regulatory elements defined | Experimental definition of cis-regulatory elements of D. melanogaster genes |
The data-type flags that were added during skim curation are stored in an internal database and used to generate a priority list for full curation. Our current practice is to target articles with the broadest range of curatable data (i.e. the highest number of flags) first. Essentially, the more flags an article has, the more relevant data types it contains and the higher its position in our priority list. The system is flexible so that we can change how the flags are used to prioritize articles as circumstances change (such as an increase in a particular type of data or funding changes). Using this approach, each individual article is prioritized based solely on its relevance to FlyBase users. In ‘full curation’, literature curators extract genetic entities and related molecular and phenotypic data from the results (text, tables and figures) and methods sections of an article. Only primary data are extracted, so no data are curated from the introduction, discussion or when they are referenced to another article. Additional types of data include GO annotations, genetic interactions (e.g. alleles of different genes), allelic interactions (e.g. alleles of the same gene), aberrations (e.g. deficiencies), allele classes (e.g. hypomorphs) and construct data (e.g. transgenic lines).
Data capture methods
Curators interact with an article in differing ways. Depending on the length of the article, some curators read the article from the computer screen, while others print the article out and highlight phrases with a marker. Often, the ‘find’ function of a PDF viewer is used to search for multiple occurrences of the same term during ‘on screen’ curation. Some curators skim through a whole article first in order to get a good overview of the work and then re-read it more thoroughly to extract the detail. Others start curation from a specific section (not necessarily the abstract or the results section) and then move to another section in search of additional information about a specific concept, for example, a particular transgenic construct or gene function. Each curator may have a different method by which they curate an article, but all curators follow strict guidelines as to what type and depth of data are curated and any inconsistencies in curation are identified in regular curator meetings.
Data from an article are added to structured template files using a range of valid symbols (and recorded synonyms), controlled vocabularies (CVs) (Table 2) and free text. These structured text file templates are known as proformae. These proformae roughly correspond to the report pages found on the FlyBase website. For genetic curation, the proformae are arranged in a hierarchy, starting with a single publication proforma, followed by gene proformae and nested allele proformae as necessary, then by proformae for other genetic entities as required (Figure 2a, proformae organization). Several proformae are bundled together to form a ‘curation record’ for each article. Curation records are opened, edited and saved in a text editor.

Literature curation into proformae. Text files composed of various proformae are used to capture data from the literature. (A) The proformae are ordered such that each curation record has to start with a publication proforma, so all objects mentioned subsequently can be attributed to the relevant publication. Allele proformae are added underneath the parent gene proforma, so all allele information can be related back to the parent gene. (B) Proformae are split into four different types of fields. The fields start with an exclamation mark (for processing) and each field has a field code, e.g. GA1a is the allele symbol field (all fields in the allele proforma are coded GAx).
The main CVs used in literature curation
| Ontology | Example search term (CV ID) |
|---|---|
| fly_anatomy | dMP2 neuron (FBbt:00001602) |
| fly_development | Pupal stage P6 (FBdv:00005353) |
| Term qualifier | Nutrition conditional (FBcv:0000714) |
| Phenotypic class | Smell perception defective (FBcv:0000404) |
| Sequence ontology | Engineered_foreign_gene (SO:0000281) |
| Origin of mutation | P-element activity (FBcv:0000486) |
| Allele class | Amorphic allele (FBcv:0000688) |
| Cellular component | Germ cell nucleus (GO:0043073) |
| Molecular function | Satellite DNA binding (GO:0003696) |
| Biological process | mRNA processing (GO:0006397) |
Each proforma is composed of a number of fields, which are split into four main types (Figure 2b, proforma fields). The first are the symbol and ID fields. These contain the unique ID and official FlyBase symbol for the genetic entity of interest. These fields also include the symbols and names used to identify the object in the literature, so users can identify an object even if FlyBase labels the object with a different symbol/name to that used in a particular article. An example of this is the gene ‘Wrinkled’, which is often identified as ‘head involution defective’ in the literature.
The second type of field houses CV lines constructed from one or more of the 16 ontologies (structured hierarchies) used in FlyBase (Table 2). CV terms are used to annotate fields for many objects in FlyBase. For example, we use Sequence Ontology (SO) (3) terms to denote the type of gene and GO terms (4) to annotate these genes for molecular function, cellular component and biological process. We also use CVs to record both the class of a phenotype and the anatomical part that is affected. Phenotypes are attached to alleles and genotypes (combinations of alleles, or alleles and chromosomal aberrations). An example piece of text from which we have extracted phenotype CV terms and free text is shown in Figure 3.
![Phenotype curation. Example data entries for a section of text [taken from an article by Baines (2003), see Ref. 5]. First, we identify the object we are ascribing the phenotype to, then we concisely curate the phenotype as free text, relating it to the object (which is placed between ‘at sign’ symbols as these symbols are hyperlinked). We then annotate the phenotype to CV terms, in this case, to terms from our ‘phenotypic class’ and ‘fly_anatomy’ ontologies.](https://oupdevcdn.silverchair-staging.com/oup/backfile/Content_public/Journal/database/2012/10.1093/database/bas039/2/m_bas039f3p.jpeg?Expires=1788067145&Signature=YKIAXV6pP51bjNxORp6N3yPPtUejA6veo62P9zcG7EOi1RKZbHQAzBBLyxlnzjiFF3v-ovQZL4KJhNs5MtJbPHr1xLuRSDg9QL4XTRrp7nV47sZ3GnwjaDid3wy~GlIhP36WHvBN~104mHxqdatyWbasYdeXLhK0lkH-FhseBI6aBSv00XqGo-esszj9gND35DaVgZZCLTdjxjQlzqeFn~EAjsHB93wGq-ERexOnhPvOnYp~ZbPOmJDi1fy75CT9lzgIDPywLoyRVXMOnPaDiBteEI6zLJoNWUPoz4-TxCSu7Ezf1C20X20rDTMGfUrn5i51yCbZMjuUgjCtP-loRQ__&Key-Pair-Id=APKAIYYTVHKX7JZB5EAA)
Phenotype curation. Example data entries for a section of text [taken from an article by Baines (2003), see Ref. 5]. First, we identify the object we are ascribing the phenotype to, then we concisely curate the phenotype as free text, relating it to the object (which is placed between ‘at sign’ symbols as these symbols are hyperlinked). We then annotate the phenotype to CV terms, in this case, to terms from our ‘phenotypic class’ and ‘fly_anatomy’ ontologies.
The third type of field is the so-called ‘SoftCV’ field, which uses a limited set of terms to describe a feature in a semi-controlled manner. The terms used are ‘structured sentences’ and do not form an ontology. An example of this type of field is the ‘Molecular lesions’ field in the allele proforma, which captures amino acid changes with the prefix ‘Amino acid replacement’.
The fourth type of field houses free text descriptions. These fields provide a level of detail that cannot be captured by CV terms and describe phenotypes and molecular lesions in a more human readable form. An example of this field is the phenotype free text field, where we record the detail of a phenotype (by paraphrasing rather than cutting and pasting from the article) that is associated with an allele or genotype (Figure 3).
Once genetic curation is complete, each curation record is checked using our in-house software, where each line is assessed for the correct structure and CV. The record is also checked for coherency between the fields, so for example, if a gene is renamed, we confirm that the new gene symbol has not been used before, and that a line is added in another field to attribute the rename to that particular article. Once a curation record has been fully checked for clashes both within the record and with the existing database, it is integrated with existing data in the FlyBase Chado database (6). Currently, we update the public website with a new release of the data six times a year, at roughly 2-month intervals.
Problems in curation
Manual curation from the literature is hard. It’s a time-consuming task that can take an experienced curator a number of hours for a standard article. It typically takes 6 months to train a FlyBase Genetic Literature Curator. Even then, curation is a social process, with curators seeking advice among the group. In this section, we will outline some of the problems we encounter in curation and describe how they could impact on the use of text mining in FlyBase.
The most common problem encountered during curation is an ambiguous genetic entity (gene, mutant allele, transgene, etc.). This situation can arise when no unique identifier (such as a FlyBase gene identifier (FBgn) or a computed gene (CG) number for genes), or an accurate and explicit reference for a mutant or transgenic line is given. Ambiguity is a particular problem when a generic symbol/name is used (e.g. ‘Actin’ or UAS-Notch), or when a symbol/name is used that is a synonym for a different entity (e.g. ‘ras’ is the current FlyBase symbol for the ‘raspberry’ gene, FBgn0003204, but is often used in the literature to refer to the ‘Ras85D’ gene, FBgn0003205). A further issue is that some symbols only differ in case-sensitivity for the first character, for example, the genes symbols ‘dl’ (dorsal) and ‘Dl’ (Delta). These ambiguities can usually be resolved by searching for associated details about the entity in the article (e.g. the use of a specific mutant allele can identify the gene being discussed) or by consulting the supplemental information for additional details. Sometimes we have to do some analysis ourselves, such as performing a BLAST search using any sequence data present in the article or supplementary files or executing an in-house script to report those entities used by a specified author in previously curated articles. As a final step, if we cannot resolve a problem, we email the corresponding author for clarification. If the ambiguity still cannot be resolved, then a curator will either associate a generic/unspecified entry for that entity with the article, or else omit the entity and add a (non-public) note to the curation record explaining the situation, with the hope that future publications will resolve the issue.
One of the more esoteric problems found in curation is the fact that multiple relationships exist between the curated data types. For example, the ‘dppEP2232 allele’ is caused by the ‘P{EP}dppEP2232 insertion’ and disrupts the ‘dpp gene’. This can cause problems for text-mining assisted curation, as the data can be attributed to the wrong object due to sentence structure or the requirement of background or contextual knowledge found in other parts of the article. In cases like this, detailed knowledge of the FlyBase proforma and curation rules, as well as a good knowledge of Drosophila biology, is necessary to ensure the correct proforma field is filled in. This is one of the reasons why we believe text-mining methods will assist manual curation rather than replace it in the near term.
Curation, therefore, is a complex process. However, when broken down into discrete components, we believe, from experience, that the use of text-mining tools can be beneficial.
Use of text-mining tools in curation
We hope that, over the coming years, we will be able to integrate text mining into multiple areas within our curation pipeline and practice, to help streamline the process. In this section, we will outline our current progress and define our aims for the near future.
FlyBase began interacting with the text-mining community in 2002 when we were involved in the KDD CUP challenge (7), a forerunner to the BioCreative initiative (8). In 2008, we developed a natural language processing (NLP) system that marked-up html versions of an article for gene/allele mentions and associated phenotypes (9). This ‘PaperBrowser’ tool, written in Java and on top of a web browser, is equipped with two navigation mechanisms called PaperView and EntitiesView. These are organized in terms of the document sectioning and possible relations between groups of words (noun phrases). More specifically, PaperView lists gene symbols (such as ‘btl’ or ‘Vang’) in the order in which they appear in each section of the article, while EntitiesView lists noun phrases related to the gene symbols such as ‘the wg pathway’. Clicking on a node in either PaperView or EntitiesView redirected the PaperBrowser tool to the sentence that contains the corresponding gene symbol or noun phrase, so allowing the curator to navigate around the article to those sections involving the entity of interest, for example, a particular mutant allele or transgene. Used in conjunction with a simple curation interface, the PaperBrowser tool improved article navigational efficiency (the number of navigation events needed before the data are extracted) by ∼58% and provided curators with enhanced utility (the number of non-navigated events needed outside of the highlighted areas) by over 74% compared to using the ‘find’ function in a PDF viewer. The PaperBrowser system is a good proof of concept, demonstrating that text mining can be successfully integrated with the FlyBase article-by-article curation system, which is something we would like to explore further in the future.
FlyBase has collaborated with WormBase (10) and Textpresso (11) since 2004 when we were consulted in the development of Textpresso for Fly, including during the creation of fly-specific vocabularies to use in the ‘Categories’ searches. In our recent collaboration, we are exploring the use of support vector machine (SVM) methods to triage primary research articles into categories based on our skim/author curation flags (12). We have trained the SVM to triage articles for new alleles, new transgenes and gene renames. We have done this through the generation of positive and negative training sets, composed of articles that we have already curated (where we know whether they contain a particular data type or not). The positive training set contains between 500 and 1000 articles, while the negative training set contains over 2000 articles. We plan to re-train the SVM for these flags periodically, to account for changes in the literature and to ensure continued low false-positive/negative rates. We are in the process of training the SVM for some of the other data triage flags, in the hope of using the SVM to triage those articles that haven’t been curated by the authors through our FTYP tool.
We can envisage text mining being used at multiple points within our curation workflow. Further to our current SVM work, text mining could be a useful adjunct to our manual GO annotation process. There are still many genes that lack functional information and, minimally, automated text mining could be used to identify articles with functional data for these genes. Following the example set by WormBase (13), we hope to use text mining to generate suggested annotations for specific genes (subject to curator review) for at least GO cellular component. We are about to embark on curating disease associations and anticipate that the Textpresso disease category could also be helpful for this aspect of curation.
While SVM methods may replicate the triage aspect of skim curation, in order to fully automate the process, we need to also identify the genes mentioned in each publication. Textpresso and the Genetics Society of America (GSA), in collaboration with WormBase, SGD (14) and FlyBase, have developed a journal article mark-up pipeline that links GSA journal articles and FlyBase gene symbols and IDs (15). False positives are discovered (at a rate of 12%, with false negatives at 27%) and resolved by a curator through a manual quality control step, with the final marked-up document assessed by the authors as part of the proofing process.
Building on our collaboration with WormBase and the GSA, we hope to use text mining to extract genetic entity symbols from all Drosophila-related literature. This, in combination with a document triage system, would form a text-mining equivalent of our skim curation, combining symbol extraction with document triaging. We are also interested in using text mining to suggest anatomy CV terms for specific genes from particular regions of text, along with gene and allele symbols and data triage flags. This would not extract the data and populate proforma, but simply highlight the text for the curator to assess and extract if appropriate. This would build on the success of the PaperBrowser system and accelerate manual curation of an article.
Summary
Over the last 20 years, FlyBase has had to adapt and change to keep abreast of changes in biology and database design. We are continually looking for ways to improve curation efficiency and efficacy. A major challenge over the years is to deal with the massive increase in data that biologists are generating, both in genomic studies and also in the published literature. While it may be too ambitious to anticipate text mining replacing human curators for full curation, we can envisage a time when text mining will be regularly used in FlyBase curation. Our aim when writing this article was to inform the text-mining community about our curation pipeline and practice. We hope that this will encourage collaboration with FlyBase so that we can put technologies in place that will aid curators both in FlyBase and in other databases to deal with this challenge.
Funding
The National Human Genome Research Institute at the National Institutes of Health [P41 HG00739] and the Medical Research Council, UK [G1000968]. Funding for open access charge: the National Human Genome Research Institute, the National Institutes of Health [P41 HG00739].
Conflict of interest. None declared.
Acknowledgements
The author thanks the BioCreative group for the invitation to give a talk at the BioCreative Workshop 2012 on the FlyBase genetic literature curation workflow, from which this article stems. He also thanks colleagues at FlyBase, particularly Steven Marygold, David Osumi-Sutherland and Gillian Millburn, for critical reading of the article, and members of FlyBase Cambridge for their work in the design of the curation pipeline and discussions on the use of text mining in curation.
References
Author notes
†The Current FlyBase Consortium comprises: William Gelbart, Nick Brown, Thomas Kaufman, Kathy Matthews, Maggie Werner-Washburne, Richard Cripps, Kris Broll, Lynn Crosby, Adam Dirkmaat, Gil dos Santos, David Emmert, L. Sian Gramates, Kathleen Falls, Beverley Matthews, Susan Russo, Andy Schroeder, Susan St. Pierre, Pinglei Zhou, Mark Zytkovicz, Boris Adryan, Marta Costa, Helen Field, Steven Marygold, Peter McQuilton, Gillian Millburn, Laura Ponting, David Osumi-Sutherland, Ray Stefancsik, Susan Tweedie, Helen Attrill, Josh Goodman, Gary Grumbling, Victor Strelets, Jim Thurmond, J. D. Wong and Harriett Platero.

{kind=link}
{kind=link}
{kind=link}