





Abstract
AcetoBase is a public repository and database of formyltetrahydrofolate synthetase (FTHFS) sequences. It is the first systematic collection of bacterial FTHFS nucleotide and protein sequences from genomes and metagenome-assembled genomes and of sequences generated by clone library sequencing. At its publication in 2019, AcetoBase (Version 1) was also the first database to establish connections between the FTHFS gene, the Wood–Ljungdahl pathway and 16S ribosomal RNA genes. Since the publication of AcetoBase, there have been significant improvements in the taxonomy of many bacterial lineages and accessibility/availability of public genomics and metagenomics data. The update to the AcetoBase reference database described here (Version 2) provides new sequence data and taxonomy, along with improvements in web functionality and user interface. The evaluation of this latest update by re-analysis of publicly accessible FTHFS amplicon sequencing data previously analysed with AcetoBase Version 1 revealed significant improvements in the taxonomic assignment of FTHFS sequences.
Database URL: https://acetobase.molbio.slu.se
Introduction
Formyltetrahydrofolate synthetase (FTHFS) is a key marker gene of the Wood–Ljungdahl pathway (WLP) of acetogenesis (1, 2). In the enzymatic process of acetogenesis, FTHFS facilitates ATP-dependent conversion of formate into formyltetrahydrofolate in the methyl branch of the WLP. Although the FTHFS gene is also present in other bacteria, e.g. syntrophic acid-oxidizing bacteria, sulphate-reducing bacteria and methanogens, only acetogens use it in the true sense of acetogenesis (1, 3–5). For extensive culture-independent investigations into the ecology of acetogenic communities in different environments, modern molecular methods are of immense importance (1). In this context, FTHFS has been used for decades as a molecular marker for the identification of potential acetogenic bacterial communities (1, 6–8). As discussed in several earlier studies, acetogens are phylogenetically diverse, metabolically very agile and important in many environments (3, 5, 9, 10). Recently, we developed a high-throughput sequencing and analysis methodology of FTHFS gene sequencing and compared it to other frequently used methods for microbial community analysis in biogas environments (4, 11). The application of our analysis strategy extensively revealed the structural composition and temporal dynamics of the potential acetogenic communities in different anaerobic digesters using different feed substrates (11, 12).
Accurate analysis of high-throughput amplicon sequencing data relies strongly on the quality and taxonomic accuracy of the reference database. For reliable identification and annotation of high-throughput amplicon sequencing data, we have created and published AcetoBase, the first curated database and repository of FTHFS sequences (3). Since its initial publication, there has been a significant increase in the number of genomic and metagenome-assembled genome (MAG) data sets containing FTHFS sequences deposited in public databases. In addition, following recent technological improvements in genomics and bioinformatics, the taxonomy of many bacterial lineages has been re-defined. Thus, for correct identification and annotation of FTHFS sequences, it is relevant and necessary to update AcetoBase with recent and updated information. In this paper, we describe recent changes and updates made to AcetoBase and present results from re-analysis of the FTHFS sequence data generated in our previous studies.
New developments
Database content
AcetoBase Version 2 contains ∼26 000 protein and ∼20 000 nucleotide sequences retrieved from public repositories. These sequences belong to 8439 distinct taxonomic identifiers. In comparison, Version 1 of AcetoBase contained ∼18 000 protein and ∼13 000 nucleotide sequences, belonging to 7 928 taxonomic identifiers. Thus, the new update has increased the AcetoBase sequence (protein and nucleotide) and taxonomic identifier diversity by ∼7 300, 7000 and 500, respectively. The major phyla associated with AcetoBase Version 2 sequences are Firmicutes, Actinobacteria, Proteobacteria and Bacteroidetes (Figure 1). An interactive version of Figure 1 is available at https://acetobase.molbio.slu.se/home/sankey. Most sequences (nucleotide and protein) in AcetoBase Version 1 were from isolated or characterized bacteria. However, in addition to sequences from bacterial isolates, in AcetoBase Version 2 FTHFS sequences from MAGs are also included. The FTHFS nucleotide and translated protein sequences belonging to MAGs do not have valid accession numbers; thus, they could not be associated with any valid identifier in National Center for Biotechnology Information (NCBI) databases. Therefore, these sequences are included as user nucleotide (UN_0000025797-UN_0000029649) and user protein (UP_0000000001-UP_0000003853) sequences in respective AcetoBase database tables while preserving complete information on the sequence origin. The FTHFS nucleotide and translated protein sequences from MAGs are included as published and new sequences, respectively, with AcetoBase as sequence owner/user identity.
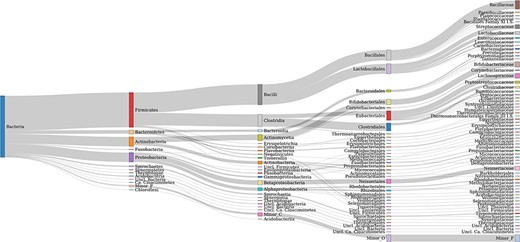
Taxonomic coverage up to the family level of FTHFS sequences present in the AcetoBase reference protein data set. Candidates with <50 sequences are merged in minor taxa.
Approximately 3100 clone sequences (47 new) are present in the AcetoBase clone data set. In AcetoBase Version 1, taxonomy prediction for the clone sequences was performed using the SINTAX algorithm (13). However, in AcetoBase Version 2, the taxonomy for the complete clone data set is predicted with the acetotax program in the AcetoScan pipeline (4) and using the updated AcetoBase reference protein data set, which is accessible at https://acetobase.molbio.slu.se/download/ref/1. Acetotax is an unsupervised sequence annotation program that filters out non-FTHFS sequences, performs best open reading frame (ORF) analysis and annotates taxonomy to FTHFS sequences based on the latest version of AcetoBase. The new taxonomic prediction of clone sequences (best ORF) indicates that most clones generated for FTHFS sequences belong to three major phyla, i.e. Firmicutes (2355/3061 sequences, 77%), Proteobacteria and Spirochaetes. The taxonomic coverage of clone sequences present in AcetoBase is presented in Figure 2. For ease of visualization, sequences (n = 2355) associated with the Firmicutes are not included in the diagram.
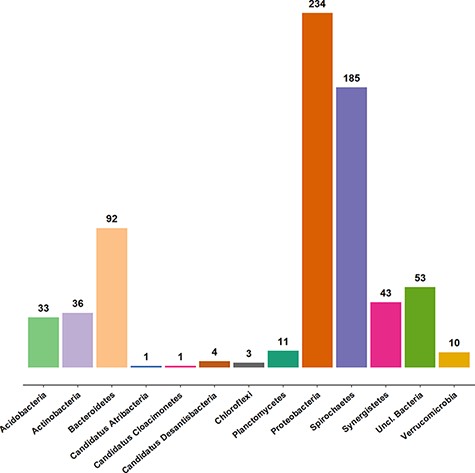
Taxonomic coverage of the clone sequence data set of AcetoBase at the phylum level, excluding Firmicutes (n = 2355 sequences). The values shown represent the sequence count for the respective phylum present in the AcetoBase clone data set.
Taxonomy prediction for the clone sequences helped in correctly associating the clone sequences to a bacterial species. For instance, it revealed that, among the sequences generated in a study by Parameswaran et al. (14), none of the sequences submitted as uncultured Alkaliphilus sp. clone (10 clone sequences) belong to Alkaliphilus. In fact, 46/47 clone sequences available from that study were found to be more closely related to Acetobacterium spp. (>94% blastx similarity) (Figure 3). These new clone sequences, with putative taxonomy and species-level percentage identity, are available in the AcetoBase clone database under accession numbers CN_0000003015–CN_0000003061. The phylogenetic trees for the reference protein, nucleotide and clone data sets have also been reconstructed according to the updated database content, and information about phylogenetic tree construction is now provided at the web interface.

Screenshot of taxonomic annotation and placement of clone sequences associated with the study by Parameswaran et al. (14) in the AcetoBase clone phylogenetic tree available at https://acetobase.molbio.slu.se/phylo/clone.
AcetoBase reference data sets
AcetoBase reference sequence databases (protein and nucleotide) were constructed from sequences present in reference tables (NP and NN) and new sequences from MAGs as described earlier (UP and UN). For the reference protein database, the NP and UP table sequences were merged, and then duplicate sequences (100% sequence similarity) were removed with the program DupRemover (15) and sequences with <300 amino acids were removed with the program FilterByLength (16). For the reference nucleotide database, the NN and UN table sequences were also merged, and then redundant sequences were removed with the program DupRemover and sequences with <1000 bases were removed with the program FilterByLength. The reference protein and nucleotide data sets in AcetoBase Version 2 contain 12 970 and 10 266 sequences and are available under the names acetobase_ref_Prot_latest.tar.gz and acetobase_ref_Nucl_latest.tar.gz, respectively. These reference databases, along with their previous versions, are available as archive databases and can be accessed at the download page https://acetobase.molbio.slu.se/download.
The latest AcetoBase data sets (protein and nucleotide) were used for the construction of phylogenetic trees by replacing invalid sequence residues with Xs in protein and Ns in nucleotide sequences using the program clean_AMINOACID_fasta (17) and clean_DNA_fasta (18), respectively. Sequences were clustered at 80% sequence similarity using VSEARCH (19) and aligned with FAMSA (20). Phylogenetic trees were then constructed with FastTree2 (21). For the construction of a clone phylogenetic tree, the taxonomically annotated clone sequence data set (described earlier), also containing both AcetoBase and NCBI accession numbers, was filtered and duplicate sequences were removed with DupRemover, remaining sequences were aligned with FAMSA and trees were constructed with FastTree2. The phylogenetic trees created (phylogenetic tree tip count: protein = 2361, nucleotide = 3941 and clone = 3048) are available on the phylogeny page. For reproducibility, the commands used for the preparation of respective trees are accompanied by interactive phylogenetic trees at the AcetoBase phylogeny web interface.
RibocetoBase data set
In AcetoBase Version 1, FTHFS sequences were connected to 16S ribosomal RNA (rRNA) genes for the respective bacteria, but these sequences were not made available as a reference data set for taxonomic annotations. In AcetoBase Version 2, we have included RibocetoBase, a 16S rRNA gene sequence data set of the FTHFS-harbouring bacterial community that contains ∼9169 sequences, which can be readily downloaded and used for taxonomic annotation of the FTHFS-containing community from 16S rRNA gene sequences. Full details of RibocetoBase and its development can be found elsewhere (11). The taxonomic coverage of the five major phyla (count >100 sequences, 8733/9169) and 13 minor phyla (count 10–100 sequences, 401/9169) are presented in Figure 4A and B, respectively. Around 20 phyla with <10 sequences (35/9169) are ignored in the visualization in Figure 4. RibocetoBase taxonomic lineages are according to the Genome Taxonomy Database (22). The RibocetoBase data set can be accessed at https://acetobase.molbio.slu.se/download/ref/3.
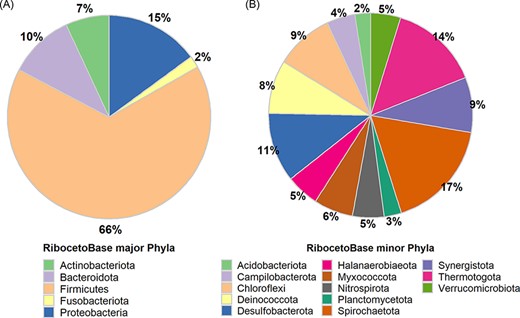
Taxonomic distribution in the RibocetoBase data set of FTHFS-harbouring communities present in AcetoBase at the phylum level. (A) Major phyla with >100 sequences and (B) minor phyla with sequence count 10–100.
Enhanced web interface and functionalities
FTHFS has long been used as a marker gene for acetogens, and several acetogens have been isolated and characterized over the years. However, a detailed inventory of the original articles and bacterial micrographs in which these acetogens were first isolated, characterized and described was lacking. In AcetoBase Version 2, we have included a bacterial micrograph collection of well-characterized acetogens (n = 52) whose FTHFS is available in AcetoBase and of acetogens proposed in the literature (n = 64), which are either not taxonomically validated or lack genome sequences. For these acetogens, links are provided in AcetoBase Version 2 to the original articles describing the isolation and description of the acetogen. A checkmark represents the original article associated with the respective acetogen, and in cases, where the original article describing the isolation and characterization of an acetogen does not have a micrograph, images from other publicly available articles have been retrieved. We have also linked the acetogen to the NCBI taxonomy database, which can be helpful in (i) getting complete information on different strains of the respective species, (ii) compiling taxonomic names and synonyms and their state of validation and (iii) linking out to other NCBI databases via the NCBI taxonomy database, for extra information. The AcetoBase Version 2 information resource for acetogens is the most descriptive and elaborate collection of acetogens to date and is accessible at https://acetobase.molbio.slu.se/organism/acetogen.
Although FTHFS is commonly used as a marker for acetogens, as mentioned earlier it is present in some syntrophic acid-degrading and sulphate-reducing bacteria (1, 3, 4). In several previous studies on FTHFS gene amplicon-based community profiling (2, 7, 8, 11, 23, 24), syntrophic acid-degrading bacteria and sulphate-reducing bacteria have been reported together with acetogenic bacteria. These groups of bacteria use the FTHFS gene in e.g., biosynthesis of folate or short-chain fatty-acid oxidation but not in reductive acetogenesis, unlike the acetogens. Thus, considering the prospects and potential for FTHFS gene profiling in different environments, in AcetoBase Version 2 we have included information (similarly to that described earlier for acetogens) on FTHFS-harbouring syntrophic acid-degrading and sulphate-reducing bacteria. It is worth noting that not all syntrophic acid-degrading bacteria and sulphate-reducing bacteria possess the FTHFS gene in their genome and that AcetoBase Version 2 only includes bacterial species harbouring the FTHFS gene. We have also grouped these bacteria according to the functional categories in which they have been described, namely syntrophic acetate-oxidizing bacteria, syntrophic propionate-oxidizing bacteria, syntrophic butyrate-oxidizing bacteria, syntrophic fatty-acid-oxidizing bacteria, sulphate-reducing bacteria and syntrophic benzene-oxidizing bacteria. This systematic grouping and collection of syntrophic bacteria can be accessed at https://acetobase.molbio.slu.se/organism/syntroph.
Search functionality and results
The new version of AcetoBase now supports searches with a query from any taxonomy level (phylum-strain) and regular expressions (matching word patterns) in the search bar. The search results are presented with a link out functionality to the respective NCBI database for genome and protein identifiers. The complete lineage for the search results is displayed with link out to the NCBI taxonomy database from the species name (Figure 5). Wherever possible, strain-level information is provided for bacterial species and MAGs. With respect to the clone data set, the search function now also supports queries with taxa names, regular expressions and searches with isolation source variables, e.g. anaerobic digester, gut, roots, colon and microbial fuel cell (Figure 5). The search results for the clone data set now also present the putative taxonomy predicted for the clone sequences, as mentioned earlier. The species-level percentage identity is available with the lineage for each clone identifier (Figure 5).

Screenshot of search results in AcetoBase Version 2 showing taxonomic lineages of the query against the nucleotide and protein data sets. The search results for the clone data set show the isolation source of the clone, its description/definition and the putative taxonomy predicted with the acetotax program.
Re-analysis of FTHFS high-throughput sequencing data
AcetoBase Version 1 was published in 2019 and was used for high-throughput sequencing data analysis in our previous studies (4, 11, 24). To evaluate the impact of the update to AcetoBase on the taxonomic annotation of the FTHFS operational taxonomic units (OTUs) generated by the AcetoScan pipeline, we re-analysed the data from our three previous studies using AcetoBase Version 2. The methodology used in the re-analysis is described in the following section.
Methodology
Re-analysis was carried out on data associated with the articles by Singh et al. (4) (data set 1), Singh et al. (11) (data set 2) and Singh et al. (24) (data set 3). These data sets were re-analysed separately with the AcetoScan pipeline, using the parameters described in the respective paper and the AcetoBase Version 2 protein data set as the reference database. The results from re-analysis of the respective data set were compared with results from previous analyses, based on differences and variations in the taxonomic annotation. For ease of understanding, in the following text, the expression ‘previous study/analysis’ refers to results published for the respective data set/paper, and ‘re-analysis’ refers to results of the re-analysis of the respective data set using AcetoBase Version 2 in the present study. Specific details of the re-analysis and major community insights gained are presented in the following sub-sections.
Re-analysis of data set 1
In re-analysis of data set 1, in addition to results from the previous study (4), we also included a new set of FTHFS sequence data for reactor GR1 that was not presented in that study, which only used data from reactor GR2 (Supplementary Figure S1A–S1F). The analysis in the original article was performed separately for the forward and reverse reads. Our re-analysis for both reactors GR1 and GR2 was conducted by merging the forward and reverse reads of each respective sample. The analysis was performed with a 100% clustering threshold and using AcetoScan analysis parameters -m 300, -n 120, -q 20, -c 5 and -t 1.0. The previous study described 935 and 662 OTUs obtained at a 100% clustering threshold from forward and reverse reads in reactor GR2, respectively. The re-analysis of data set 1 and the new data for reactor GR1 resulted in 1025 OTUs at a 100% clustering threshold when using merged data from forward and reverse reads. The re-analysis results indicated a higher percentage of annotated sequences compared with the previous analysis and with no unclassified OTUs at the genus level (Supplementary Figure S1E). Genera related to OTUs that were not classified in the re-analysis, but were classified in the previous analysis, were Butyrivibrio, Caldisalinibacter, Eggerthella, Hungateiclostridium, Lagierella, Maledivibacter and Phocea. Taxa classified in the re-analysis but not classified in the previous study included, e.g., Peptoniphilus, Ruminiclostridium, Sedimentibacter, Thermoanaerobacter and Urinicoccus. The genus Senegalimassilia was only observed in the new additional data from reactor GR1 and not in the data from reactor GR2 used in the previous study (Supplementary Figure S1E).
Re-analysis of data set 2
Re-analysis of data set 2 was done for forward reads as described in the original article (11), with the AcetoScan analysis parameters -m 300, -n 150, -q 21, -c 10, -r 1, -e 1e-30 and -t 1.0. The number of OTUs (n = 387) generated in the re-analysis was similar to that generated in the previous study (n = 391). The re-analysis results (Supplementary Figure S2A–S2F) for data set 2 at the phylum level showed a significant reduction in the annotation of OTUs classified previously as Actinobacteria and, in contrast to the previous analysis, no annotations were obtained for Spirochaetes at relative abundance (RA) >1% (Supplementary Figure S2A). Compared with the previous study, the fraction of OTUs classified as Firmicutes increased on re-analysis. At the genus level, genera that were not classified in the re-analysis were Caloranaerobacter, Methylocystis, Oscillibacter, Phocea, Tissierella, Treponema and Varibaculum (Supplementary Figure S2E). Clear and significant differences in the taxonomic annotations were noted for the OTUs previously annotated as Varibaculum, which in the re-analysis were annotated as unclassified Urinicoccus. The total number of genera (RA >1%) annotated in the re-analysis was 17, compared with 21 in the previous study. Apart from the changes at the genus level, the most significant change in the re-analysis was for species belonging to the phylum Cloacimonetes (Supplementary Figure S2F). In the re-analysis, only one species from this phylum was classified, i.e. Candidatus Cloacimonetes bacterium (MAG ID: AS05jafATM_99) originating from anaerobic digesters (25). In comparison, the previous study annotated two species, Cloacimonetes bacterium HGW-Cloacimonetes-1 and Cloacimonetes bacterium HGW-Cloacimonetes-2, which originated from a metagenomics project analysing subsurface sediments (26).
Re-analysis of data set 3
Data set 3 was generated in a study by Singh et al. (24) involving high-throughput microbiological surveillance of six different biogas plants (one plug flow and five parallel continuous stirred-tank reactors plants) with a total of 11 biogas reactors. The AcetoScan analysis parameters used in the re-analysis were the same as those used in the previous study (-m 300, -n 150, -q 20, -c 5, -e 1e-30 and -t 1.0). The re-analysis resulted in 1899 OTUs, compared with 1901 OTUs in the previous study (Supplementary Figure S3A–S3F). At the phylum level, the re-analysis results revealed similar community diversity in all reactors as previously reported, except for reactor C6. The re-analysis results showed an increase in the percentage of OTUs annotated as Firmicutes, with a corresponding decrease in OTUs for the phylum Actinobacteria. A minor reduction was observed for OTUs previously belonging to Synergistetes, which were not classified as Synergistetes in the re-analysis (Supplementary Figure S3A). Due to the high numbers of genera in both the previous study and the re-analysis, the results obtained for each genus and species are not included here (see Supplementary Figures S3E and S3F for comparison). However, an overview of the annotation results indicated that annotation accuracy improved significantly in the re-analysis, with a smaller number of resulting taxa and higher percentage identity to the reference database than in the previous study.
Discussion
The FTHFS sequence data generated in previous studies were re-analysed here using the updated Version 2 of AcetoBase, and differences in taxonomic annotation were compared. Careful interpretation of the re-analysis results and cross-comparison to the previous studies (4, 11, 24) supported and justified the improvements in taxonomic annotations resulting from the updates to AcetoBase. The increased taxonomic diversity in the reference database was shown to provide an opportunity for taxonomic annotation of OTUs with higher accuracy (greater percentage identity and lower e-value) in the best-hit strategy used by AcetoScan. However, since FTHFS is not a taxonomic marker and due to technical limitations in FTHFS amplicons sequencing, the community diversity evaluated with the FTHFS gene can have similarities, but also differences, when compared to the 16S rRNA gene and whole-genome metagenome-based community profiles. Nevertheless, a comparative analysis of FTHFS and 16S rRNA gene amplicons by Singh et al. (11) demonstrated that the microbial community profiled with FTHFS showed high similarity to the structure and dynamics revealed by community profiling using the 16S rRNA gene. The recent changes in the taxonomic lineages of bacteria also contributed to differences between the re-analysis results and those in the previous studies.
Scope of acetobase
Acetogenic bacterial communities are metabolically diverse and are found in hugely varied environments such as anaerobic digesters, animal gut, rumen, human gut, marine sediments, forest soil, peatlands, permafrost and microbial electrochemical systems (10, 11, 14, 24, 27–34). In these environments, they are not only involved in the carbon cycling via acetate but are also extensively involved in the metabolism of ethanol, methanol, formate, butyrate, lactate, vanillate, chlorinated compounds, cellulose, mono-tetra saccharides and methylated amines (2, 35–43).
AcetoBase was initially developed for use in the analysis of the acetogenic community in anaerobic digestor/biogas environments. However, AcetoBase contains large numbers of FTHFS-harbouring bacterial species that are also present in other natural environments. As discussed earlier, FTHFS primers can target syntrophic acid-oxidizing, sulphate-reducing bacterial groups, etc., but also different fermentative FTHFS-harbouring bacteria. These fermentative bacteria play important roles in various anaerobic environments and are also of great physiological importance for humans and animals, including Bacteroidetes, Bifidobacterium, Clostridium, Eubacterium, Faecalibacterium and Lactobacillus (44–47). AcetoBase thus has great potential to be a representative database for FTHFS-based microbial ecological analyses in varied environments. For instance, the human gut harbours large numbers of acetogenic bacterial communities that are significantly involved in gut physiology, even in the presence of methanogens and sulphate-reducing bacteria (39, 48). Furthermore, members of acetogenic or FTHFS-possessing communities, namely Clostridium, Blautia, Eubacterium, Eggerthella, Prevotella, Ruminococcus and family Lachnospiraceae, are reported to be substantially involved in e.g. dysbiosis of autoimmune disorders, multiple sclerosis, rheumatoid arthritis, systemic lupus erythematosus, cirrhosis, gastrointestinal disorders and Parkinson’s disease (49–53). Despite this, only a few studies have examined the FTHFS-harbouring communities in the human gut (54, 55), while any large-scale longitudinal study on this subject is completely lacking.
Conclusions
This paper describes recent updates to AcetoBase in terms of database content, user interface and functionality and presents results from re-analysis, using the updated Acetobase (Version 2), of FTHFS sequence data published in our earlier studies. The updated database content includes additional sequences from new bacterial species and MAGs. This increase in taxonomic diversity is intended to enable FTHFS community profiling and taxonomic annotation with higher accuracy. The updates to the web interface, especially for acetogens and syntrophic acid-oxidizing and sulphate-reducing bacteria, may allow it to serve as a knowledge bank of acetogenic and syntrophic organisms that possess the FTHFS gene. The improved functionality can facilitate searches of the database and retrieval of information on the taxonomic lineage of different bacterial species. The putative taxonomy provided for clone sequences can be of significant help in determining the correct taxonomy, which can differ from the taxonomic associations of published clone sequences, or in future FTHFS clone library-based studies.
The re-analysis of FTHFS sequence data from our previous studies demonstrated and supported the usefulness of the database update in improving the taxonomic annotations resulting from AcetoScan analysis. The variations in taxonomic annotations obtained using the updated AcetoBase Version 2, compared with Version 1, did not change the overall dynamics or interpretations of community profiles. Most of the changes in taxonomic annotations were observed at lower taxonomic levels and among members of the same order or family. The addition of new sequences from MAGs helped improve the identification of FTHFS sequences (higher percentage similarity and lower e-value). As amplicon sequence analysis is dependent on reference databases, continuous updates to these databases are needed and the future addition of new sequences may further improve taxonomic annotation. AcetoBase Version 2 contains sequences from acetogens and syntrophic organisms. Detection of this acetogenic and syntrophic composite employing FTHFS amplicon sequencing could open up new avenues in ecology and enable functional studies of microbial interactions in different environments, especially anaerobic bioprocesses and the gut of animals and humans. Ecological analyses and understanding of syntrophic microbial interactions are currently scarce, so longitudinal FTHFS profiling and faster analyses of community dynamics could be an outstanding tool in gaining important insights into the unknown acetogenic and syntrophic microcosm.
Supplementary data
Supplementary data are available at Database Online.
Acknowledgements
We would like to thank Hans-Henrik Fuxelius for his support and help in the original code modification and developing Version 2 of AcetoBase.
Funding
Swedish University of Agricultural Sciences.
Conflict of interest
None declared.
Data availability
The FTHFS raw sequence data from reactor GR1 analysed in this study and associated with our previous study (4) have been submitted to NCBI SRA (study: SRP336508) under BioProject accession number PRJNA761914. The OTU sequences generated in the re-analysis of data sets 2 and 3 have been submitted to AcetoBase under accession numbers UN_0000023501-UN_0000023887 and UN_0000023888-UN_0000025796, respectively.
Author contributions
A.Si. is involved in the conceptualisation, data curation, methodology, software, visualization and writing—original draft. A.Sc. is involved in the conceptualisation, funding acquisition, resources and writing—review & editing.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}