





Abstract
We introduce SilkBase as an integrated database for transcriptomic and genomic resources of the domesticated silkworm Bombyx mori and related species. SilkBase is the oldest B. mori database that was originally established as the expressed sequence tag database since 1999. Here, we upgraded the database by including the datasets of the newly assembled B. mori complete genome sequence, predicted gene models, bacterial artificial chromosome (BAC)-end and fosmid-end sequences, complementary DNA (cDNA) reads from 69 libraries, RNA-seq data from 10 libraries, PIWI-interacting RNAs (piRNAs) from 13 libraries, ChIP-seq data of 9 histone modifications and HP1 proteins and transcriptome and/or genome data of four B. mori-related species, i.e. Bombyx mandarina, Trilocha varians, Ernolatia moorei and Samia ricini. Our new integrated genome browser easily provides a snapshot of tissue- and stage-specific gene expression, alternative splicing, production of piRNAs and histone modifications at the gene locus of interest. Moreover, SilkBase is useful for performing comparative studies among five closely related lepidopteran insects.
Database URL: https://silkbase.ab.a.u-tokyo.ac.jp
Introduction
The silkworm Bombyx mori is the only fully domesticated insect that has been used for silk production for >5000 years (1). In addition to its industrial applications, B. mori is a model insect in genetics, molecular biology, physiology and pathology. For instance, Kametaro Toyama reported the Mendelian inheritance of the cocoon color of B. mori (2), which was the discovery that Mendelian laws are verified in animals. Currently, B. mori is used for producing a large amount of a single protein via genetic engineering (3) or baculovirus vectors (4).
The draft genomes of B. mori were independently constructed and reported by Chinese and Japanese groups in 2004 (5, 6), which were merged and assembled with newly obtained fosmid- and BAC-end sequences to form a 432-Mb-long new genome in 2008 (7). However, this genome assembly (ver. 2008) still contains various gaps primarily due to a huge number of repetitive sequences within the genome. To solve this problem, our group performed re-sequencing of B. mori genome by PacBio and Illumina sequencing platforms and obtained a new genome in 2016 with a total length of 460.3 Mb (8). The new genome assembly (ver. 2016) and newly predicted gene models (ver. 2017) were stored and made available in SilkBase.
SilkBase was developed in 1999 as the B. mori expressed sequence tag (EST) database. The first version of SilkBase contained about 35 000 ESTs from 36 complementary DNA (cDNA) libraries (9). Subsequently, several B. mori databases have been released, for instance, SilkDB (https://silkdb.bioinfotoolkits.net) (10), KAIKObase (https://kaikobase.dna.affrc.go.jp) (11), Silkworm Base (https://shigen.nig.ac.jp/silkwormbase/top.jsp), SilkPathDB (https://silkpathdb.swu.edu.cn) (12) and SGID (http://sgid.popgenetics.net) (13). Some of them contained our genome assembly (ver. 2016) and gene models (ver. 2017) (11, 13), whereas SilkDB has been updated by replacing the genome assembly and gene models (ver. 2008) (7) with other ones (10). The SilkDB includes genome datasets that were made from our group’s PacBio sequence reads (8), transcriptome, Hi-C and the genome data from 163 different geographically representative strains (10). The KAIKObase is the B. mori’s genome database that includes the genome assembly and gene models (ver. 2017), genetic maps and lists of manually curated gene families for pesticide targets and silk proteins (11). The SGID is a comprehensive and interactive database containing the genome assembly (ver. 2016) and gene models (ver. 2017). The genome browser in the SGID provides domestication levels at each gene locus by comparing sequences of B. mori and its putative ancestor Bombyx mandarina (13). In addition to these B. mori databases, several lepidopteran genome databases, such as the lepidoDB (https://bipaa.genouest.org/is/lepidodb/), lepbase (http://lepbase.org) (14), KONAGAbase (http://dbm.dna.affrc.go.jp/px/) (15) and MonarchBase (http://monarchbase.umassmed.edu) (16) are currently available.
As described above, SilkBase was originally established as the EST database. This EST database has been updated several times as an integrated database for B. mori transcriptome and genome resources and stably maintained availability for ‘23 years’. In this paper, we introduce the status of SilkBase that provides researchers quick and reliable outputs from accurate datasets using useful and comfortable in-built tools and browsers.
Materials and methods
Construction of sequence data
The de novo assembly of RNA-seq reads was performed using Trinity (17). The open reading frames (ORFs) of the RNA-seq assemblies were predicted using TransDecoder, which is the plugin of Trinity (17). Hypothetical genomes were constructed by substituting different nucleotides from the genome assembly (ver. 2016) using BWA (18), SAMtools (19) and GATK (20). Furthermore, genome sequences of B. mandarina (Sakado strain) were obtained using Illumina HiSeq 2500 and were then assembled using Platanus (21) with fosmid-end sequences.
Data annotation
The gene models (ver. 2017) were annotated using InterProScan (22) and blastp search against NCBI’s non-redundant (nr) protein data sets. The transcript levels [transcripts per million (TPM)] of each gene model (ver. 2017) in RNA-seq libraries were estimated using Bowtie 2 (23) and original R scripts. Gene ontology (GO) terms of each RNA-seq assembly were determined using ncbi-blastp against UniProtKB/Swiss-Prot. The transcript levels (TPM and fragments per kilobase of exon per million mapped fragments) of the RNA-seq assemblies were estimated using RSEM (24).
Data construction for the genome browser
The 2016 version of the genome assembly was used as the genome for the following data construction. Sequences of RNA-seq assemblies, cDNA reads, gene models (ver. 2008) and gene set A (25) made in 2013 were mapped to the genome using GMAP (26). The location of genes on chromosomes was determined in the process of gene prediction (8) and was then used for mapping gene models (ver. 2017) to the genome browser. Next, RNA-seq reads were mapped to the genome using HISAT2 (27). In addition, PIWI-interacting RNA (piRNA)- and ChIP-seq reads were mapped to the genome using Bowtie (28) with no mismatch and multimap. ChIP-seq peak calling was performed using epic2 (29).
Web interface and server construction
The Web interface was written in HTML, CGI, JavaScript and Perl. MySQL was used for the database management, and JBrowse (30) was used for the genome browser.
Data source
Bombyx mori
BAC clones derived from BAC-end sequences, BAC-derived assemblies (31) and W chromosome-derived BAC sequences (32). Hypothetically reconstructed genome and raw read of B. mori 18 strains derived from C108T, N4, b20, c10, c51, d18, e10, f35, g53, k25, n16, o55, o56, p20, p21, p22, p44 and u48. RNA-seq libraries designated as anterior silk gland, brain, early embryo, epidermis, fat body, internal genitalia, midgut and middle silk gland of B. mori p50T strain (8), early embryo of B. mori N4 strain and epidermis of B. mori otm mutant strain (33). cDNA-end reads derived from cDNA libraries and designated as an--, bmmt, BmN, bmnc, bmov, bmte, br--, brP-, brS-, caL-, ce--, ceN-, cesb, e40 h, e96 h, epV3, F1mg, famL, fbpv, fbS2, fbVf, fbVm, fcaL, fcP8, fdpe, fe100, fe8d, fepM, ffbm, FJsb, fmgV, fmxg, fner, fphe, fprw, ftes, fufe, FWD, fwgP, heS0, heS3, J150, JFsb, maV3, MFB, mg--, msgV, N--, Nnor, NRPG, NV02, NV06, NV12, ovS0, ovS3, P5PG, pg--, prgv, ps4M, psV3, tesS, tesV, vg4M, wdS0, wdS2, wdS3, wdV1, wdV3 and wdV4 (9). piRNA libraries designated as OV, TE, MW, WF, LY, Siwi, BmAgo3, GFP#8, 0h Egg, 6h Egg, 12h Egg, 24h Egg and 40h Diapaused Egg (32, 34–36). ChIP-seq reads designated as Input, pIZ, BmHP1a, Cdp1, IgG-R, H3K4me1, H3K4me2, H3K4me3, H3K9ac, H3K9me2, H3K9me3, H3K27ac, H3K27me3, H3K36me3, IgG-M and Pol2 (37–39). All information about the data source can be obtained from the library information page of the B. mori on SilkBase.
Bombyx mandarina
Hypothetically reconstructed genome and raw read of B. mandarina two strains derived from Sakado and Oki strains. RNA-seq libraries designated as B. mandarina anterior silk gland, midgut and middle silk gland. All information about the data source can be obtained from the library information page of the B. mandarina on SilkBase.
Trilocha varians
RNA-seq libraries derived from Trilocha varians (40) antennae (female), antennae (male) and midgut. All information about the data source can be obtained from the library information page of the T. varians on SilkBase.
Ernolatia moorei
RNA-seq library derived from Ernolatia moorei (40) midgut. This information can be obtained from the library information page of the E. moorei on SilkBase.
Samia ricini
RNA-seq libraries derived from Samia ricini anterior silk gland, midgut and middle silk gland. cDNA ends derived from S. ricini fat body and embryo (41). All information about the data source can be obtained from the library information page of the S. ricini on SilkBase.
Results
Data content
To avoid ‘Garbage In, Garbage Out’, unreliable data in public databases were not installed in SilkBase. Our group or collaborators obtained most data used in our database, particularly next-generation sequencing data. Some of these data have been published in peer-reviewed scientific journals (Table 1).
A summary of the datasets installed in SilkBase
| Category | Library | Description | References |
|---|---|---|---|
| Bombyx mori | |||
| Genome | Chromosome-level genome assembly | 28 chromosomes and 668 scaffolds | (8) |
| Fosmid library | 274 342 fosmid clonesb | ||
| BAC libraries | 137 753 BAC clones from three libraries | (31, 32) | |
| Hypothetically reconstructed genome | 782 316 in total from 18 strainsb | ||
| Re-sequenced genome DNA libraries | 1 672 128 940 reads from 18 strainsb | ||
| Old scaffold library (2008) | 43 462 scaffolds | (7) | |
| Gene | Gene model | 16 880 genes | (8) |
| Transcript level, protein family membership, domains and repeats, detail signature matches, residue annotation, GO term prediction and description against nr | |||
| Gene model (2008) | 14 623 genes | (7) | |
| Position on genome, GO term prediction, Uniref and Orthologs | |||
| Geneset A | 16 823 genes | (25) | |
| Position on genome | |||
| Transcriptome | Assembled RNA-seq libraries | 1 062 486 in total from 10 tissues/stagea | (8) |
| Position on genome, GO term prediction, description against nr and transcript level | |||
| ORF from assembled RNA-seq libraries | 529 531 in total from 10 tissues/stagea | (8) | |
| Position on genome, GO term prediction and description against nr | |||
| RNA-seq libraries | Reads from 10 tissues/stage | (8, 33) | |
| The complete sequences of the FL-cDNA clone libraries | 11 833 clones | (25) | |
| Position on genome | |||
| cDNA libraries | 461 119 in total from 69 libraries | (9) and unpublished | |
| Position on genome, GO term prediction, Uniref and Orthologs | |||
| SAGE libraries | 82 227 clones | (42) | |
| MPSS libraries | 44 872 clonesa | ||
| piRNA libraries | 83 984 212 reads in total from 13 libraries | (32, 34–36) | |
| Transposon libraries | 121 of well-annotated transposons and 1690 of transposons | (43) | |
| Epigenome | ChIP-seq libraries | 526 234 147 reads from 16 libraries | (37–39) |
| Peak calling | |||
| Bombyx mandarina | |||
| Genome | Scaffold-level genome assembly | 66 797 scaffoldsb | |
| Fosmid libraries | 153 216 clonesa | ||
| Hypothetically reconstructed genome | 86 924 in total from two strainsb | ||
| Re-sequenced genome DNA libraries | 185 322 718 reads from two strainsb | ||
| Transcriptome | Assembled RNA-seq libraries | 141 139 in total from three tissuesa | |
| GO term prediction, description against nr and transcript level | |||
| ORF from assembled RNA-seq libraries | 73 790 in total from three tissuesa | ||
| GO term prediction, Description against nr | |||
| Trilocha varians | |||
| Transcriptome | Assembled RNA-seq libraries | 106 248 in total from three tissuesa | |
| GO term prediction, description against nr and transcript level | |||
| ORF from assembled RNA-seq libraries | 41 707 in total from three tissuesa | ||
| GO term prediction and description against nr | |||
| Ernolatia moorei | |||
| Transcriptome | Assembled RNA-seq libraries | 38 954 assemblya | |
| GO term prediction, description against nr and transcript level | |||
| ORF from assembled RNA-seq libraries | 15 068 ORFsa | ||
| GO term prediction and description against nr | |||
| Samia ricini | |||
| Genome | Scaffold-level genome assembly | 155 scaffolds | (44) |
| Transcriptome | Assembled RNA-seq libraries | 171 159 in total from three tissuesa | |
| GO term prediction, description against nr and transcript level | |||
| ORF from assembled RNA-seq libraries | 78 839 in total from three tissuesa | ||
| GO term prediction and description against nr | |||
| cDNA | 20 320 clones from two libraries | (41) | |
| GO term prediction, Uniref and Orthologs | |||
This database.
This database (Sequenced by National Bio Resource Project).
Bombyx mori
SilkBase contains B. mori genome assembly (ver. 2016), which was constructed by our group (8). In addition, it contains information about 274 342 fosmid clones, 137 753 BAC clones, hypothetically reconstructed genome and 1 672 128 940 raw reads (>QV30) of the genomes of 18 strains and 43 462 scaffolds of the genome assembly (ver. 2008) (7). Moreover, it comprises 16 880 gene models (ver. 2017) (8), 14 623 gene models (ver. 2008) (7) and 16 823 gene set A (26). The gene models (ver. 2017) were annotated with transcript levels, GO terms, blast results against nr and InterProScan results. Transcriptome data include 1 062 486 de novo assemblies of RNA-seq reads, 529 531 putative ORFs predicted from RNA-seq assemblies and RNA-seq raw reads. The RNA-seq de novo assemblies and predicted ORFs linked with genome loci, GO terms, blast results against nr and transcript levels. In addition, the following were installed: 11 833 complete sequences of the full-length cDNAs (26), 461 119 cDNA-end reads (9), 82 227 tags of serial analysis of gene expression (SAGE) (42), 44 872 signatures of massively parallel signature sequencing (MPSS) and 83 984 212 reads of piRNA libraries (32, 34–36) with 121 well-annotated and 1690 predicted transposons (piRNA precursors) (43). Epigenetic data include 526 234 147 reads of ChIP-seq (37–39). All information about these datasets can be obtained from the library information page of the B. mori on SilkBase.
Bombyx mandarina
SilkBase contains 66 797 genome scaffolds and 153 216 fosmid-end reads of Sakado strain of B. mandarina. It also contains a hypothetically reconstructed genome and 185 322 718 raw reads (>QV30) of the genome of two strains. A total of 141 139 de novo assemblies of RNA-seq reads and 73 790 ORFs predicted from RNA-seq assemblies were also installed. RNA-seq assemblies and predicted ORFs were annotated with GO term prediction, descriptions against nr and transcript levels (RNA-seq assemblies only). All information about these datasets is available on the library information page of the B. mandarina on SilkBase.
Trilocha varians
A total of 106 248 de novo assemblies of RNA-seq reads with GO terms, blast results against nr and transcript levels were installed. Additionally, 41 707 ORFs predicted from RNA-seq assemblies with GO term predictions and descriptions against nr were installed. All information about these datasets is available on the library information page of the T. varians on SilkBase.
Ernolatia moorei
A total of 38 954 de novo assemblies of RNA-seq reads with GO terms, blast results against nr and transcript levels were installed. Furthermore, 15 068 ORFs predicted from RNA-seq assemblies with GO terms and blast results against nr were installed. All information about these datasets is available on the library information page of the E. moorei on SilkBase.
Samia ricini
A total of 155 scaffolds of S. ricini genome assembly (44), 171 159 de novo assemblies of RNA-seq reads, 78 839 ORFs predicted from RNA-seq assemblies and 20 320 cDNA ends (41) were installed. The RNA-seq assemblies and predicted ORFs were annotated with GO terms, blast results against nr and their transcript levels (RNA-seq assemblies only). All information about these datasets is available on the library information page of the S. ricini on SilkBase.
Simple graphical user interface
The graphical user interface of SilkBase (Figure 1) is configured with the top page, blast search pages, the piRNA- and ChIP-seq mapping tool, keyword search pages, result pages, library information pages and the genome browser page. Direct links to all the main features, which are separated for each species, are displayed on the top page. On the blast search pages, a homologous sequence search is available. The piRNA- and ChIP-seq mapping tool is used for mapping piRNA- and ChIP-seq reads in the dataset of B. mori. Keyword search is also available for all species. All the SilkBase datasets are listed in a table format on the library information page, and most of them are linked to the detailed information page that also directs the genome browser. The transcriptome, genome and epigenome data are available in an integrated format on the genome browser.
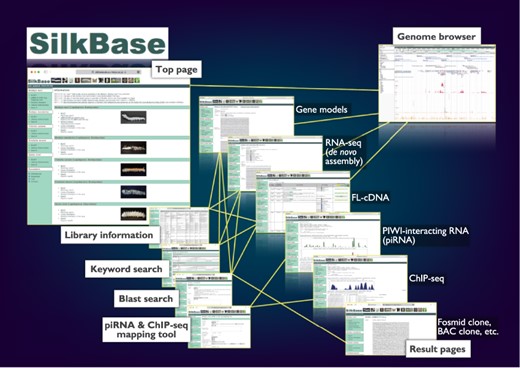
Overview of the Web user interfaces. The path lines show the user pathways between the Web user interfaces.
Integrated genome browser
SilkBase users can access the genome browser from the top page or each gene (clone) page of the website. On this genome browser, a snapshot of tissue- and stage-specific gene expression (RNA-seq and cDNA ends), alternative splicing (RNA-seq and full-length cDNAs (FL-cDNAs)), piRNA production (piRNA-seq) and histone modifications (ChIP-seq) at the gene locus of interest on B. mori genome and gene models (Figure 2A) is also available.
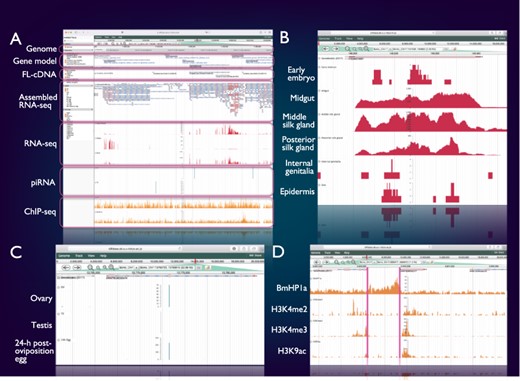
Integrated genome browser. (A) Overview of the genome browser. Genomic, transcriptomic and epigenomic information is displayed on a single screen. (B) Transcriptional status of BmSuc1 in different tissues. (C) A piRNA-producing locus in the Masc gene. (D) Histone modifications around the KWMTBOMO06377 gene.
Figure 2B shows one of the results of RNA-seq mapping on the genome browser. The BmSuc1 encodes a functional β-fructofuranosidase, which is specifically expressed in the midgut and silk gland (45). This tissue-specific expression of BmSuc1 is clearly seen on our genome browser: RNA-seq reads from midgut, middle silk gland and posterior silk gland are abundantly mapped onto the BmSuc1 locus, whereas few reads are mapped in the RNA-seq libraries of the early embryo, internal genitalia and epidermis (Figure 2B).
Figure 2C shows an example of a piRNA-producing locus within the protein-coding gene. Masculinizer (Masc) encodes a protein required for masculinization and dosage compensation in B. mori. Our studies revealed that Masc messenger RNA is depleted by the W chromosome-derived Feminizer (Fem) piRNA in females and Masc also produces a Masc-derived piRNA via a ping-pong cycle (46–48). We can verify this result clearly on the genome browser: Masc piRNA can be seen in piRNA-seq libraries of ovary and 24-h post-oviposition egg but not in those of testis (Figure 2C).
Figure 2D shows a snapshot of histone modifications at a certain genome locus around KWMTBOMO06377. The ChIP-seq reads of three euchromatic marks, i.e. H3K4me2, H3K4me3 and H3K9ac (37), are abundantly mapped onto the gene body of KWMTBOMO06377. However, ChIP-seq reads of HP1a (B. mori heterochromatin protein 1 homolog), which is associated with the transcription start sites (TSSs) of highly expressed genes (38), can be seen at the TSS of KWMTBOMO06377. Combining with RNA-seq data, we can easily understand the transcriptional and epigenetic conditions of any gene on the B. mori genome.
piRNA- and ChIP-seq mapping tool
SilkBase users can examine the piRNA production status of any query sequence graphically and identify abundance, location and sequences of corresponding piRNAs (Figure 3A). Figure 3B shows a piRNA mapping result of Fem, which indicates a single abundant piRNA (Fem piRNA) produced from the sense strand. This tool was used to visualize Fem- or transposon-derived piRNAs in our previous studies (46–48). We can also use the ChIP-seq mapping tool on SilkBase because it provides abundance, location, and sequences of ChIP-seq reads (HP1 and histone marks) against any query sequence (Figure 3C).
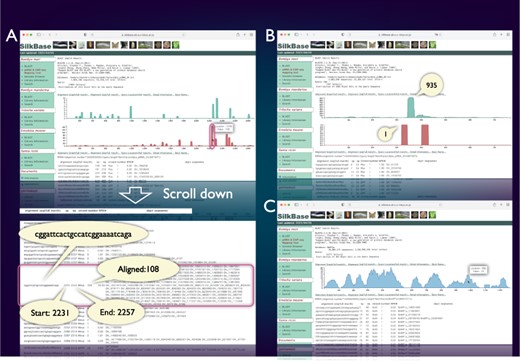
piRNA- and ChIP-seq mapping tool. The vertical axis indicates the nucleotide position of the query sequence, and the horizontal axis indicates the depth of coverage. The upper graph shows the mapping results on the sense strand, whereas the lower graph shows those on the antisense strand (piRNA mapping tool only). Information on nucleotide sequence, start point, end point, strand and the depth of coverage is displayed below the graph. (A) piRNA production in ovary from the transposon Kabuki. The detailed information of a certain peak (indicated by oval) is as follows: the piRNA sequence is ‘cggattcactgccatcggaaaatcaga’, and 108 reads are mapped from 2231 to 2257. (B) An example of the piRNA production status in the Fem locus. The depth of coverage is 935 on the sense strand and 1 on the antisense strand. (C) Mapping of the HP1a-binding sites around the KWMTBOMO02692 gene. The depth of ChIP-seq coverage on both strands is merged and displayed on a single graph (ChIP-seq mapping tool).
Comparative genomic analysis of B. mori-related species
SilkBase comprises the transcriptome and/or genome resources of B. mori-related species, B. mandarina (Lepidoptera: Bombycidae), T. varians (Lepidoptera: Bombycidae), E. moorei (Lepidoptera: Bombycidae) and S. ricini (Lepidoptera: Saturniidae) (Figure 4A). Bombyx mandarina is a putative ancestor of B. mori and is commonly found in mulberry fields in East Asia. Meanwhile, T. varians is widely distributed in South and Southeast Asia, and its larvae feed on the leaves of Ficus spp. Ernolatia moorei is found in South and Southeast Asia, and its larvae feed on the leaves of Ficus spp. Furthermore, S. ricini (Eri silkmoth), a gigantic and polyphagous saturniid moth, is the almost fully domesticated saturniid species. The genome sequence of these species was recently determined by our collaborators (44).
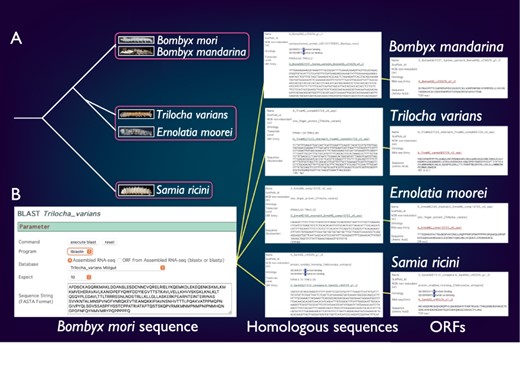
Comparative genomic analysis of B. mori-related species. (A) The phylogenetic relationship of B. mori and related species. The host plants are different among the three groups (indicated by rectangles). (B) An example of the identification of B. mori Masc homologs from four species.
SilkBase users can perform comparative genomic analysis between B. mori and these species. Figure 4B shows an example of the identification of B. mori Masc homologs. The users can obtain the homolog sequences by a tblastn search using the amino acid sequence of the B. mori Masc as a query against transcriptome assembly of B. mori-related species at each species tab (49). In addition, the ORF of each homolog sequence is available from the result page.
Conclusions
SilkBase is an integrated database of B. mori and related species. It consists of B. mori’s newly assembled chromosome-level genome, B. mori’s transcriptome and epigenome data generated from highly reliable reads of next-generation sequencers and four related species’ transcriptome and/or genome data, most of which were obtained and assembled in our laboratory. The unique selling points of SilkBase are as follows: (1) the simple graphic interface and powerful server provide researchers with good-looking results quickly, and the snapshots of results can be readily used for figure preparation, (2) researchers can easily understand the situation of tissue- and stage-specific gene expression, alternative splicing, piRNA production and histone modifications at a glance on our genome browser and analytic tools and (3) SilkBase provides a platform for conducting comparative studies among five closely related lepidopteran insects. In conclusion, SilkBase is a user-friendly database and we hope that researchers in the world routinely use this database for their studies on B. mori and other insects.
Acknowledgements
We thank Toru Shimada, Kazuei Mita, Shinpei Kawaoka, Katsuhiko Ito, Keisuke Shoji and Masaru Tamura for their helpful comments and discussions. Computational resources for the annotation of assembled RNA-seq were provided by the Data Integration and Analysis Facility, National Institute for Basic Biology.
Funding
Grant-in-Aid for Publication of Scientific Research Results, JSPS, Japan (1999-2003, 2005-2018 to Toru Shimada).
Conflict of interest
None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}