





Abstract
Three-dimensional domain swapping is a unique protein structural phenomenon where two or more protein chains in a protein oligomer share a common structural segment between individual chains. This phenomenon is observed in an array of protein structures in oligomeric conformation. Protein structures in swapped conformations perform diverse functional roles and are also associated with deposition diseases in humans. We have performed in-depth literature curation and structural bioinformatics analyses to develop an integrated knowledgebase of proteins involved in 3D domain swapping. The hallmark of 3D domain swapping is the presence of distinct structural segments such as the hinge and swapped regions. We have curated the literature to delineate the boundaries of these regions. In addition, we have defined several new concepts like ‘secondary major interface’ to represent the interface properties arising as a result of 3D domain swapping, and a new quantitative measure for the ‘extent of swapping’ in structures. The catalog of proteins reported in 3DSwap knowledgebase has been generated using an integrated structural bioinformatics workflow of database searches, literature curation, by structure visualization and sequence–structure–function analyses. The current version of the 3DSwap knowledgebase reports 293 protein structures, the analysis of such a compendium of protein structures will further the understanding molecular factors driving 3D domain swapping.
Database URL:http://caps.ncbs.res.in/3dswap
Introduction
Protein structures are elementary units of form and function in living organisms. Structural properties of proteins can be comprehensively explained using the concept of primary, secondary, teritiary and quaternary structures (1–5). Proteins accomplish their specific functions by interacting with a wide variety of micro- and macromolecules within the cell. Some of these interactions are mediated by oligomerization in proteins (6–10). In general, protein oligomers are formed by the polymerization of protein monomeric subunits into dimers or higher order oligomers. Protein oligomers can be broadly classified into two classes: homo-oligomers (formed by the oligomerization of identical subunits), and hetero-oligomers (formed by the oligomerization of non-identical subunits). Several key molecular functions rely on molecular interactions facilitated by such protein oligomers with other molecular players in the cell (11–13).
3D domain swapping is a protein structural phenomenon where two or more protein chains form a dimer or higher oligomers by exchanging an identical structural element between the monomers (14–16). Several native (natural/physiological) intramolecular interactions within the monomeric structures are replaced by intermolecular interactions of protein structures in swapped oligomeric conformations (17). Although the term ‘3D domain swapping’ was initially defined to describe the structure of diphtheria toxin, a similar structural phenomenon was predicted over four decades prior to that, during experiments with dimers of ribonuclease (RNaseA) with partly knocked-out active sites (18–22). 3D domain swapping was proposed as an important mechanism to explain the evolution of proteins from monomeric to oligomeric conformations to mediate a specific function. The presence of distinct ‘hinge’ and ‘swapped’ regions is the hallmark of protein structures in 3D domain swapping conformation. Structures in swapped conformations were reported to perform a variety of functions, and proteins involved in deposition diseases (like neurodegenerative diseases, amyloidosis and Alzheimer's disease) have been reported in 3D domain swap conformations (23–28). Several mechanisms have been proposed to explain oligomeric formation and protein aggregation (29–31). Most of these prior studies were focused on a limited number of protein molecules. Specific experimental and computational studies to understand the molecular mechanisms behind 3D domain swapping proposed various features like macromolecular crowding (27), protein concentration (32), evolutionary constraints, mutational effect on residues in hinge regions (32, 33) and changes associated with pH (27, 34) to be associated with the phenomenon. It has been observed in a variety of protein structures that belong to different folds, source organisms, protein domain families and diverse secondary structural elements. However, the biological implications and structural basis of 3D domain swapping still remain elusive. The first, curated database on 3D domain swapping, ‘3DSwap’ is a useful resource that can be analyzed to answer specific questions pertaining to sequence, structure and functional implications of 3D domain swapping. Curated data and functional annotations compiled in 3DSwap can be used to develop algorithms that can predict various aspects (e.g. hinge and swapped regions) from sequence or structure data and enable the comparative analysis of higher order residue interactions and various structural properties.
Earlier studies on 3D domain swapping were focused on understanding the phenomenon from experimental (35–45) and computational perspectives (46–54), but no integrated database or bioinformatics resource has been developed based on it. The collective study of 3D domain swapping in proteins, by compiling a large data set of proteins reported with this mechanism, will be an important step toward understanding the various factors that control this phenomenon, from the molecular level to its crucial role in deposition diseases and other functions mediated by swapping (K. Shameer and R. Sowdhamini, unpublished results). The catalog of proteins reported in 3DSwap database was generated using an integrated pipeline of database searches, literature curation, structure visualization and subsequent analysis.
Features of 3D domain swapping
Several earlier studies defined different structural and functional aspects of 3D domain swapping. To generate a generic framework to understand swapping in protein structures, various key terminologies like ‘bona fide domain swapping’; ‘quasidomain swapping’, ‘swapped region’ and ‘hinge region’ were introduced earlier (27, 34). Protein structures involved in 3D domain swapping are classified into two major groups (15, 34): bona fide domain swapping and quasidomain swapping (55). Bona fide domain swapping refers to structures formed where while both the monomer and dimer of a molecule exist in stable forms, the dimer adopts a domain swapped conformation and the monomer adopts a closed conformation. Examples of protein structures in bona fide domain swapping category include the diphtheria toxin (18, 34), RnaseA (16, 56), cluster of differentiation 2 (CD2) involved in T-cell adhesion (57), stage 0 sporulation protein A (spo0A) involved in the regulation of sporulation response (58), etc. Quasidomain swapping refers to the structures formed when some proteins form domain swapped oligomers without a known closed monomer. If these proteins have homologs known to be closed monomers, these oligomers are considered to be ‘quasidomain swapped’. Examples of quasidomain swapped protein structures include crystalline (59), pheromone/odorant binding/transport proteins (60) crystallin (59), pheromone/odorant binding/transport proteins (60), RYMV (viral capsid protein) (61) and human cystatin C (protease inhibitor) (62). An example of a bona fide domain swapped protein structure is shown in Figure 1 (PDB ID: 11BG) and a quasidomain swapped protein structure is provided in Figure 2 (PDB ID: 1CKS). The two basic features of a structure undergoing this phenomenon are the swapped and hinge regions. The swapped region is defined as a globular domain or a short structural element that is intertwined with other protein chains in an oligomeric conformation. The hinge region is the short stretch of amino acids, mostly in a loop conformation, that links the swapped region and the remaining core of the protein. Hinge and swapped regions in bovine seminal ribonuclease are highlighted in Figures 1 and 2. The swapped region could be a small loop in some proteins or a whole domain in some others. Hence, we introduce a set of four new terms, ‘primary inter-domain interface’, ‘secondary inter-domain interface’, ‘secondary minor region’, and ‘secondary major region’ (as defined in Methods section) to quantify swapping with respect to the whole structure, using a measure called ‘extent of swapping’ (ES).
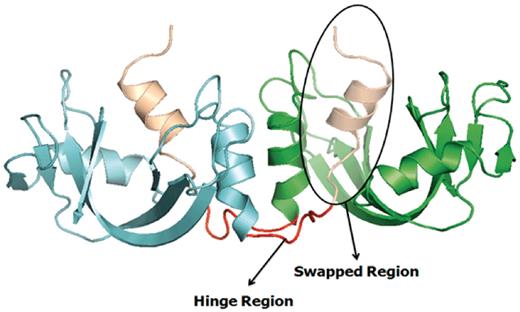
Example of a bona fide domain swapping structure: bovine seminal ribonuclease (PDB ID: 11BG) in 3D domain swap conformation with highlighted hinge and swapped regions. Individual chains are colored in cyan and green. Hinge regions are colored in red and swapped region is colored in coffee brown.
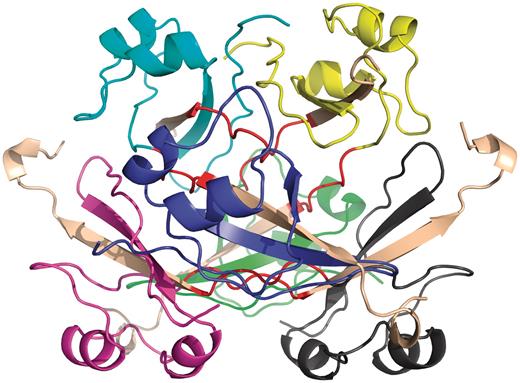
Example of quasidomain swapping structure: hexameric assembly of human CksHs2 (PDB ID: 1CKS). Six individual chains are colored in different colors (yellow, cyan, violet, blue, green, gray). Hinge regions are colored in red and swapped region is colored in coffee brown.
Methods
The 3DSwap database has been developed using a multi-step literature curation approach coupled with structure analysis, structural mapping and integration of function annotation.
Concept of secondary interface regions in structures with 3D domain swapping
We define ‘primary inter-domain interface’ as the link between domains in the monomeric form of the protein. The primary interface is the native interface in monomer, primarily mediated by intermolecular interactions, which are replaced by intramolecular interactions due to 3D domain swapping. ‘Secondary inter-domain interface’ is defined as the additional interface present in the domain swapped dimer, but not in the monomer. The entire domain or a part of a protein chain that is swapped with the other domain of the neighboring chain is referred to as the ‘secondary minor region’ or ‘guest’. The region that interacts with the secondary minor region is termed as the ‘secondary major region’ or ‘host’. A graphical account of various structural features of 3D domain swapping is illustrated in Figure 3.
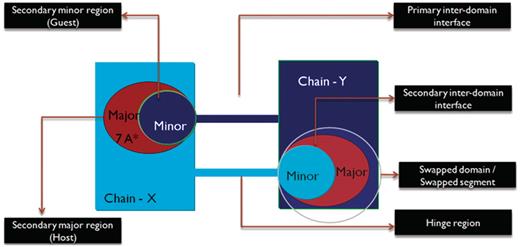
Schematic representation of the different features of 3D domain swapping.
Extent of Swapping (ES)
where RMI = number of residues in secondary major interface, Rmi = number of residues in secondary minor interface. Searches within a 7-Å distance shell were performed around Cα atoms of all residues in secondary minor interface (Rmi) to retrieve residues in secondary major interface (RMI) using a custom FORTRAN script (see Figure 3 for the schematic representation of various structural features). RChain:X = residues in chain X and RChain:Y = residues in chain Y. Based on the ES values, protein structures in 3DSwap are classified into three major classes as ‘extensive swapping’, ‘moderate swapping’ and ‘minimum swapping’. Thresholds were defined using the twilight zone concept from sequence–structure homology (63, 64). We hypothesize that if an oligomeric structure is affected by >35% of its entire length, there may be significant structural impacts due to domain swapping. Extensive swapping, therefore, refers to the oligomeric structures where ES is observed to be within the range of 35–100%. Moderate swapping refers to the oligomeric structures where ES is observed to be within the range of 15–35% and minimum swapping refers to the oligomeric structures where ES is observed to be <15%. Different classes of ‘ES’, with representative examples are given in Figure 4 [extensive swapping: 63.87%—1DXX (65); moderate swapping: 27.64%—1O4W (66); and minimum swapping: 8.5%—1BL9 (67)]. A large number of proteins were observed to be in the ‘extensive swapping’ category (Figure 5), suggesting that the impact of 3D domain swapping on the oligomeric conformation is significant in the majority of the swapped proteins. Further analysis is required to ascertain the correlation between the ES and several structural classes.
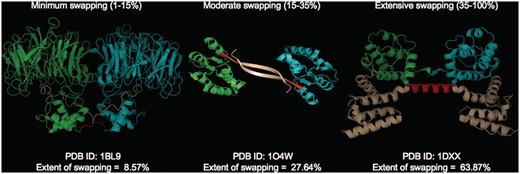
Different classes of ‘ES’ with representative examples (PDB IDs: 1BL9,1O4W and 1DXX). Individual chains are colored in cyan and green. Hinge (red) and swapped regions (brown) are mapped in different colors.
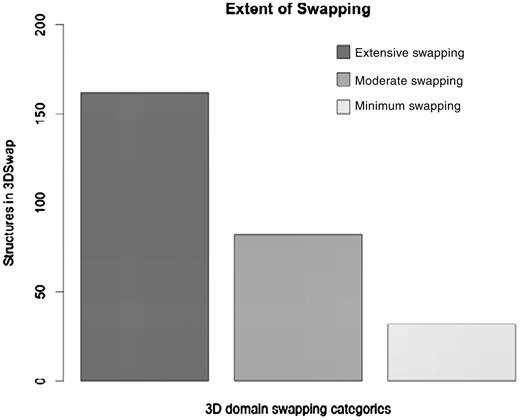
Distribution of protein in the data set based on ES.
Literature-based protein structural curation method
We combined a literature-based structural information mining and manual structural visualization approach in an iterative fashion to identify and map features of 3D domain swapping to structures. We define this approach as ‘literature-based protein structural curation’. The initial list of entries were obtained using text mining searches in the advanced search interface of the Protein Data Bank (PDB) (68). We used the basic keywords related to 3D domain swapping (‘domain-swap’, ‘domain-swapping’, ‘3D domain swap’, ‘3D domain swapping’) and their derivatives for the initial search in the primary database PDB. Using the basic search utility, we obtained an initial pool of PDB identifiers (PDB ID) and their respective PubMed identifiers (PMID) that reported the keywords. Similar searches were performed in PubMed and IDs retrieved for the PubMed entries with these set of keywords in their title, abstract or full-length research articles. PMIDs were further mapped to PDB IDs using the PDB Advanced Search Interface. An index list of non-redundant PDB IDs and their respective PMIDs were generated. For each pair of IDs, the respective PDB files were downloaded and the full-length literature article obtained as available. The literature data that reported the structure determination methods and biochemical properties of the proteins were used as a guide to identify the hinge and swapped regions in protein structures.
Database searches were performed using ‘domain swapping’ and its derivatives, and a list of entries made. ‘Keyword’ searches were performed against the PDB and PubMed databases. Furthermore, these entries were manually checked using derived data provided in the PDB (69), PDBSum (70) and the PQS database (11). Structures were also assessed using the Domain Identification Algorithm (DIAL server) (71, 72) to understand the structural domain architecture and boundaries of the proteins. Structures of confirmed 3D domain-swapped cases were visualized and analyzed for various 3D domain swapping structural features using PyMOL (The PyMOL Molecular Graphics System, Version 1.2r3pre, Schrödinger, LLC.). Furthermore, literature curation was performed to obtain details about the residues in the hinge and swapped domains. Confirmed structural entries were processed to obtain various features. All features of a protein structure with well-defined hinge and swapped regions were extracted and reported in the database.
Biological and biomedical text mining approaches are useful in identifying putative relationships between various entities like genes, proteins, small molecules and diseases defined in the literature using automated methods (73, 74). Such automated text mining approaches were not suitable for the curation of 3D domain swap-related features. It is a fairly straightforward task to identify the initial list of protein structures involved in 3D domain swapping using the ‘PDB Advanced Search Interface’ and subsequent structural visualization. But, the challenge in developing 3DSwap was in the structural mapping of the residues in hinge and swap region to the protein structures involved in swapping. Information about the hinge or swapped region or the respective location of these regions was not available in the abstract. In most cases, this was provided in the main text, figure legend or in tables and automated text mining of the abstract became impossible. Furthermore, the mapping of residues in hinge and swapped regions was challenging due to several known issues associated with the raw data deposited in the PDB. For example, chain break, missing residues, re-numbering of residues and missing ATOM records, often hinder one-to-one mapping of information from the literature to the protein structures. Due to such reasons, in many cases, the number of residues mentioned in the literature did not directly match with the structures and hence manual mapping of the residues with the help of visualization tools was required for the curation of hinge and swapped regions.
In summary, an entry in the 3DSwap knowledgebase, as per the workflow, starts with a valid PDB ID. The PDB ID is used to obtain the key literature associated with the structure based on the PubMed ID integrated in the PDB. Literature is manually reviewed to identify the residues in the hinge and swapped regions. These residues are identified and mapped on to the structure with the help of visualization tools. Furthermore, the sequence and structure were used to compile various features reported in the database. Ambiguous cases were removed and several structures with incomplete information on the hinge or swapped regions were not included in the current version of 3DSwap. Various steps associated with the curation of protein structures included in 3DSwap are summarized in the flow chart (Figure 6).
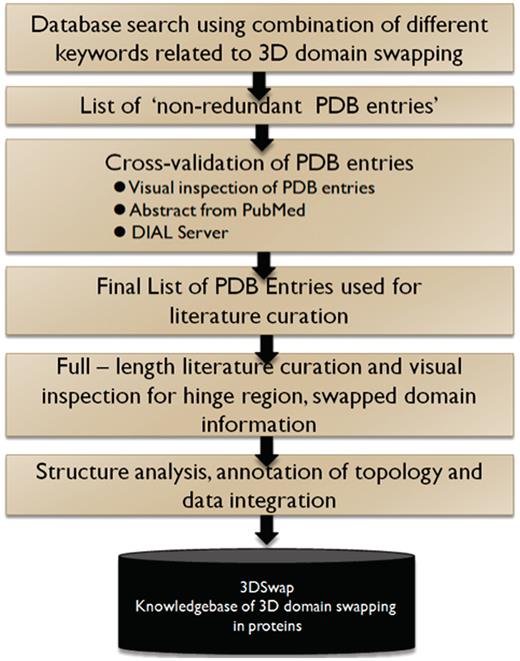
A schematic representation of curation steps involved in the development of 3DSwap knowledgebase.
Tools used for literature-based protein structural curation
Literature-based protein structural curation was performed by using the following tools and databases: PDB (68), PQS Server (11), PDBe (75), PDBSum (70), PyMOL (76), RasMol (77, 78) and DIAL (71, 72). The PDB, PQS Server and PDBe were used to obtain the structural coordinates in PDB format, the quaternary structural coordinates and the biological assembly in oligomeric form. PyMOL and RasMol were used for the visualization of structures to confirm 3D domain swapping and mapping of the hinge and the swapped regions from literature. The DIAL server was used to identify the architecture of structural domain definitions of the proteins. The information obtained for the list of proteins involved in 3D domain swapping, after literature-based protein structural curation and structural bioinformatics analysis, has been compiled into a new knowledgebase ‘3DSwap’. After the data curation steps (Figure 6), various structural and sequence features are calculated for the entries reported in 3DSwap.
Technical details
The web interface of 3DSwap knowledgebase was developed using HTML and JavaScript. Perl-CGI programs were used for the development of search, query and retrieval systems. Back-end data is stored using MySQL. Scripts for parsing PDB and accessory files, search tools and calculation of various features were coded in Perl. JOY package was used for the calculation of various structural features. SEQPLOT was developed using Perl and amino acid indices derived from AAINDEX database (79) were used to generate the plots. Functional patterns in the hinge region, swapped region and full sequence were identified by scanning the sequence of protein structure involved in swapping using ScanProsite (80) with PROSITE (81) data downloaded on November 2010.
Results
Protein structures reported with 3D domain swapping include a variety of folds, source organisms, families and diverse primary and secondary structures.
Search and browse utilities in 3DSwap
Users can browse within 3DSwap using ‘Full list of entries in 3DSwap’, which provides a dynamic table of protein structures available in the current version of 3DSwap. Users can sort the tables in 3DSwap database using the column headers. Users can also browse the 3DSwap knowledgebase using structures categorized into three different groups according to ES values. Screenshots of browse interfaces are provided in Figure 7.
Browse interfaces of 3DSwap.
The search interface of 3DSwap provides a convenient approach to access the database based on searches using keywords, metadata from PDB (PDB ID and PDB header), SCOP domain, SCOP fold, Pfam domain, GO annotation and CDD annotations. The Basic Local Alignment Search Tool (BLAST) (82) based sequence search interface is provided to search the database for full-length proteins involved in domain swapping, hinge region and swapped domains. This will enable the user to query the 3DSwap knowledgebase using sequence data and retrieve the sequence homologs. The BLAST interface is designed to perform a search against three distinct databases: database of full-length sequences, hinge and swapped regions. Screenshots of search interfaces are provided in Figure 8.
Search interfaces of 3DSwap.
Interactive visualization of swapped segments and hinge regions
Molecular visualization tools play an important role in the structural analysis of protein molecules. To support interactive visualization and analysis of protein structures involved in domain swapping, we have provided options for protein visualization using Jmol (83) and RasMol (84). Jmol is a Java-based web applet that enables the users to visually analyze the molecular structures within a web browser. The Jmol applet loaded with a structure can be obtained from the Jmol logo. RasMol is a standalone molecular visualization application, which can be installed on different operating systems. We provide RasMol scripts for the interactive analysis of structures in local machines. A screenshot is provided in Figure 9 with Jmol-based visualization for PDB ID 11BG. Hinge and swapped regions are highlighted for visual exploration of such residues.
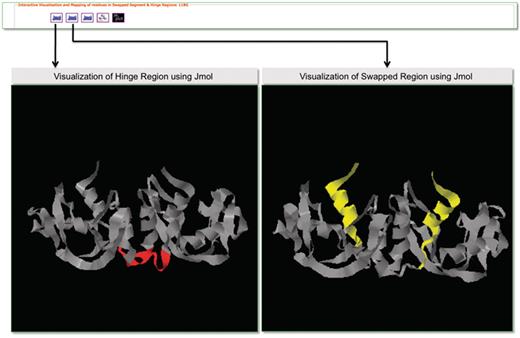
Screenshot that highlights visualization in 3DSwap using Jmol.
2D Plots: annotated topology diagrams
We obtained 2D secondary structural topology diagrams from PDBSum; these topology diagrams are annotated to define hinge and secondary minor regions. Such a 2D diagram can be utilized for the quick visualization of hinge regions, swapped regions and secondary major interfaces to understand the effect of swapping on the whole protein structure.
Amino acid plots
Amino acid plots refer to a collection of bar charts derived from the amino acid composition of protein sequences within hinge and swapped regions and full structures of proteins involved in 3D domain swapping. These profiles will provide a preview of the composition of residues involved in hinge and swapped regions. These profiles will be a useful option to compare the composition between the different regions. The ‘pepstats’ program from EMBOSS package is used to calculate statistics of protein properties.
Structural features reported in 3DSwap
Hydrogen bonds, solvent accessibility, secondary structure and JOY-based representations were obtained using JOY package (85) for all the entries in the database. JOY files are also provided for every entry reported in the database. A parseable text file (with extension .tem) generated by the JOY package is also provided for full oligomeric structure, hinge and swapped regions. This file provides various structure-derived data [secondary structure and phi angle, solvent accessibility, hydrogen bond to main chain CO, hydrogen bond to main chain NH, hydrogen bond to other side chain/heterogen, cis-peptide bond, hydrogen bond to heterogen, covalent bond to heterogen, disulfide, main chain to main chain hydrogen bonds (amide), main chain to main chain hydrogen bonds (carbonyl), DSSP (86), positive phi angle, percentage accessibility and Ooi number]. Accessory files, based on structural features, are provided to enable the user to utilize the database and files for further analysis. A screenshot that depicts the various structural features compiled in 3DSwap is provided in Figure 10.
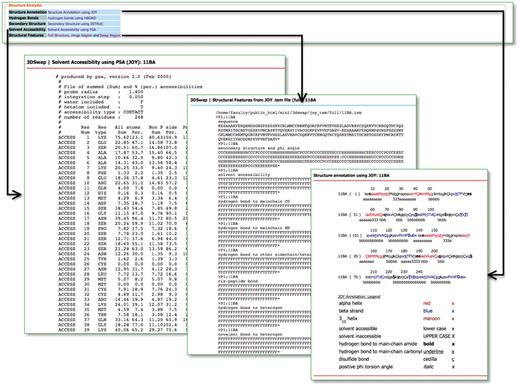
Structural features in 3DSwap.
Function annotation
Function annotation information derived from the Gene Ontology Annotation (GOA) (87) and Pfam databases (88) [using structure-based function annotation data from the SIFTS initiative (89)] are provided. Function annotation data are provided as cross-reference links to retrieve all entries in 3DSwap. For example, users can click on a particular GO ID or Pfam ID to view all protein structures with that particular annotation from 3DSwap. To depict the functional diversity of proteins in 3DSwap, we mapped the PDB IDs in 3DSwap to GO IDs using SIFTS resource, and the frequency of each GO IDs were calculated for biological process categories and visualized in treemap using REVIGO (90) (Figure 11). The treemap indicates the diversity of biological processes mediated by the proteins in the current version of 3DSwap. Annotation data was cross-referenced with Pfam (88), AmiGO (91) and CDD databases (92–94) to retrieve further information about the protein domains or GO terms.
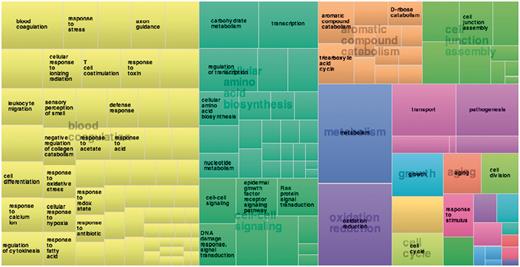
Treemap derived using frequency of proteins annotated in 3DSwap using biological process category.
Sequence analysis in 3DSwap
The sequence analysis section of 3DSwap provides two sets of features. SEQPLOT enables users to generate plots of any three of the various amino acid indices, reported in Amino Acid Index (AAINDEX) database (95, 96), in a single window. AAINDEX is a database of various amino acid physicochemical properties, substitution matrices and statistical protein contact potentials. Five hundred and sixteen amino acid indices are integrated in the current version of SEQPLOT. SEQPLOT will help users to understand the various physicochemical properties of amino acids encoded in different regions (hinge, swap and full sequence) of the protein. The ‘PROSITE pattern’ link in the 3DSwap knowledgebase provides information about the functional motifs associated with the full sequence, hinge and swapped regions identified using the ScanProsite (80, 97) algorithm. This option will help the user explore functional motifs present in various regions of proteins involved in 3D domain swapping.
Bovine seminal ribonuclease: description of an example structure from 3DSwap
To illustrate various features of the 3DSwap knowledgebase, we discuss various features using the example of bovine seminal ribonuclease, a hydrolase enzyme from Bos taurus (PDB ID: 11BG). The screenshot of the 3DSwap page for 11BG is provided in Figure 12.
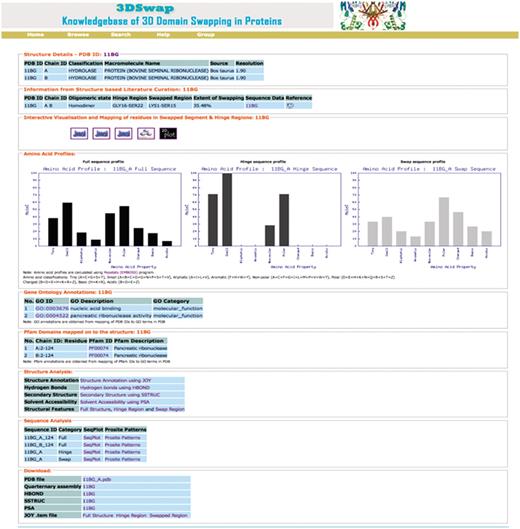
Screen shot of 3DSwap knowledgebase for the PDB ID: 11BG.
The first section of the 3DSwap database provides literature-curated information about 3D domain swapping from the literature and the public structural bioinformatics and function annotation databases. 11BG (56) is the structure of Bovine seminal ribonuclease protein with 3D domain swapping observed between two chains (A, B). ‘Biological unit’ refers to the oligomeric state of a protein that can form a biologically active unit. The oligomeric state of 11BG is ‘homodimer’ and is provided in 3DSwap, which are derived from the PQS and PDBe databases (75). Information about the residues in the hinge and swapped regions is obtained, starting from the literature curation, followed by the careful inspection of structures. For example, in 11BG [44], seven residues (GLY16-SER22) are involved in the hinge region. In 11BG, 15 residues (LYS1-SER15) are observed in the swapped segment. It is classified as a member under the ‘extensive swapping’ class of proteins since the ES value for this dimer is 35.5%. A link to access sequence data provides various structure-derived sequence data, including hinge region (secondary minor region), secondary major region, swapped region and full sequence data in FASTA format. Links to access interactive visualization tools for analysis of protein structures involved in 3D domain swapping are provided. GO annotations are available for 11BG in two categories: molecular function (nucleic acid binding, catalytic activity, nuclease activity, endonuclease activity, pancreatic ribonuclease activity, hydrolase activity) and cellular component (extracellular region). A single pancreatic ribonuclease domain (RnaseA domain, Pfam ID: PF00074) is present in individual chains of 11BG. Amino acid profile plots of hinge and swapped regions, and full protein sequence data based on PepStat is given in the ‘Amino acid profiles’ section, which is followed by output from various structure analysis tools and download files.
3DSwap—database statistics
The current version of 3DSwap contains 293 structures, as of November 2010, with literature curated and information integrated about 3D domain swapping and annotations derived from GO, Pfam and SCOP. A total of 293 structures were mapped to a total of 4099 GO annotations. These include 59 unique GO terms in the cellular component category, 244 unique terms in the biological process category and 139 unique terms in the molecular functions category. A total of 826 Pfam domain annotations are reported in the current version of 3DSwap with 175 unique Pfam domains. From a structural perspective, the entries in the current version of 3DSwap were mapped to 757 SCOP entries with representative members from 107 SCOP folds, 127 SCOP superfamilies, 151 SCOP families and 176 SCOP domains (Figure 13).
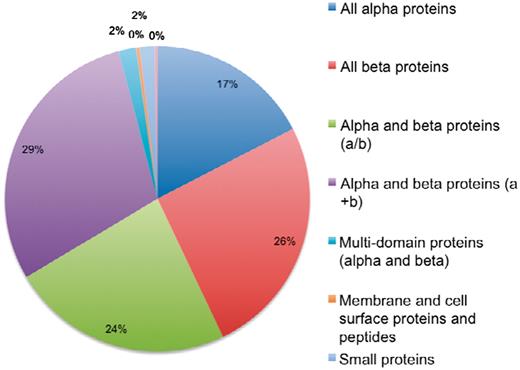
Distribution of protein structures in 3DSwap in SCOP classes.
Coverage of various annotations incorporated in the current version of 3DSwap is as follows: the current version of 3DSwap contains CDD annotations for 97.66% of the protein chains of protein structures compiled in 3DSwap. CDD annotations are derived using individual protein chains of proteins structures compiled in 3DSwap. A total of 729 protein chains were used to query the CDD database and 712 protein chains were annotated with atleast one CDD domain. SCOP annotations could be retrieved for 238 structures (81.22%), Pfam annotations were available for 278 structures (94.88%) and GO annotations were available for 196 structures (66.89%).
Discussion
The PDB is a primary resource among various protein structural databases available to the structural bioinformatics community. Several secondary databases are developed based on protein structural coordinates derived from the PDB. 3DSwap is a secondary database developed using protein structural data derived from the PDB and is developed to collect and document various features of proteins involved in 3D domain swapping. Biocuration (98, 99) is gaining significant importance in biology due to the need for integrating multiple data resources, coupled with literature evidence and bioinformatics-based predictions, to understand new molecular connections which are not obviously visible without such integration. Curated databases have made a significant contribution to the dissemination of biological knowledge by compiling data into publicly available databases that enabled enhanced interpretation and knowledge based inference of biological data (100–106). We believe curation efforts like 3DSwap can add more knowledge to the available knowledge arena in biology and can be highly benefited from open access scholarly publications. The database statistics of protein structures in 3DSwap mapped to SCOP classes indicate that 3D domain swapping structures are reported in eight different SCOP classes (Figure 13). No structures are reported in the current version of 3DSwap that belong to coiled-coil proteins, low-resolution structures and peptide classes in SCOP. The curation strategy ‘literature-based protein structural curation’, used to compile protein structures in 3DSwap can be applied as a generic protein structure curation strategy for structural curation studies in the future.
Curated protein databases like the Conserved Domain Database (CDD) (94) which builds alignment search models guided by 3D structure and SCOP are extremely useful for understanding structure, function and the classification aspects of protein families. The literature-curated data compiled in the 3DSwap database, can be utilized for in-depth analysis of various aspects of 3D domain swapping. For example, we have used the sequences of proteins reported in 3DSwap in our earlier attempts to predict 3D domain swapping using features derived from structure and sequence (107) and an algorithm to predict 3D domain swapping from sequence information using Random Forest (108). We have also utilized the data for a detailed meta-analysis of proteins involved in 3D domain swapping and to understand the functional role of swapped proteins (K. Shameer and R. Sowdhamini and K. Shameer, Prashant S. and R. Sowdhamini, manuscripts in preparation). We envision that the 3DSwap knowledgebase and its features, such as integrated visualization tools and the various sequence and structure analysis results, will benefit the structural bioinformatics community, and enable researchers to perform comprehensive analysis and classification of proteins involved in 3D domain swapping.
The curated data and the sequence, structure and function data compiled in 3DSwap will enable large-scale analysis of 3D domain swapping; for example, users can download and utilize the solvent accessibility to analyze the percentage of residues that are accessible in various regions of protein oligomers involved in 3D domain swapping. Thus, we believe 3DSwap will emerge as the primary data resource to obtain several new insights into the sequence, structure and functional aspects of 3D domain swapping.
Future work and updates
Since proteins involved in the 3D domain swapping phenomenon play a prominent role in the structural, functional and regulatory aspects, future versions of 3DSwap will be released periodically, depending upon the relative availability of a significant number of structural entries with 3D domain swapping in the PDB. As manual curation is required for identification and mapping of hinge and swapped regions, an automatic update is currently not possible. However, we trust this manuscript will be followed by a community effort to record such valuable data as domain swapping in the abstracts of publications that report protein structures involved in swapped conformations. This will greatly enable automation in future. The current version of the 3DSwap database is focused on developing a primary resource that documents various primary features of 3D domain swapping. Secondary features, like the classification of structures involved in 3D domain swapping into quasi-domain or bona fide swapping and information about homologs, will be introduced in the future updates of 3DSwap. It should also be noted that the current version of 3DSwap contains only representative structures from various SCOP classes, which are involved in 3D domain swapping, from multiple SCOP superfamilies and CDD database superfamilies, and does not comprehensively cover those superfamilies. Further initiatives will be taken to integrate the data from 3DSwap to various structural bioinformatics resources and biological wikis like GeneWiki (109), PDBWiki (110) and WikiPathways (111). Incorporation of the curated data into protein sequence, structure and function-related biological wikis will improve the accessibility and usage of literature-curated data compiled in 3DSwap. Efforts will also be taken up to expand the annotation and cross-references to other important protein sequences, structure and function-based databases like PDBSum (70), GeneWiki (109), WikiPathways (111), CATH (112), STRING (113), SMART (114) and BioGPS (115) in the future updates.
Conclusion
We developed a curated knowledgebase of proteins involved in 3D domain swapping provided in the public domain. The literature mining approach employed to identify features of 3D domain swapping in proteins can be modified and the curation strategy applied in different contexts of protein structural curation. The current version of the 3DSwap database provides various sequence, structure and functional features of proteins involved in 3D domain swapping. A quantitative measure, ‘ES’, is defined to quantify the magnitude of swapping in an oligomeric structure. We envisage that the availability of the 3DSwap database, with its curated content and information related to domain swapping, will be one of the most useful resources for the research community interested in studying 3D domain swapping in great detail.
Funding
National Centre for Biological Sciences (TIFR) and Department of Biotechnology, India, to R.S. and K.S; Wellcome Trust, UK, Senior Research Fellowship to R.S. Funding for open access charge: National Centre for Biological Sciences (TIFR)
Conflict of interest. None declared.
Acknowledgements
R.S. and K.S acknowledge the National Centre for Biological Sciences (TIFR) for infrastructural and the Department of Biotechnology, India, for financial support. R.S. was a Senior Research Fellow of the Wellcome Trust, U.K. We thank the anonymous referees for their constructive suggestions and valuable comments.
References
Author notes
Present address: Khader Shameer, Division of Cardiovascular Diseases, Mayo Clinic, Rochester, MN 55905, USA

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}