





Abstract
Large-scale RNA interference (RNAi) experiments, especially the ones based on short-interfering RNA (siRNA) technology became increasingly popular over the past years. For such knock-down/screening purposes, different companies offer sets of oligos/reagents targeting the whole genome or a subset of it for various organisms. Obviously, the sequence (and structure) of the corresponding oligos is a key factor in obtaining reliable results in these large-scale studies and the companies use a variety of (often not fully public) algorithms to design them. Nevertheless, as the genome annotations are still continuously changing, oligos may become obsolete, so siRNA reagents should be periodically re-annotated according to the latest version of the sequence database (which of course has serious consequences also on the interpretation of the screening results). In our article, we would like to introduce a new software/database tool, the RNAiAtlas. It has been created for exploration, analysis and distribution of large scale RNAi libraries (currently limited to the human genome) with their latest annotation (including former history) but in addition it contains also specific on-target analysis results (design quality, side effects, off-targets).
Database URL:http://www.rnaiatlas.ethz.ch
Introduction
Recently, RNA interference (RNAi), a natural mechanism for gene silencing (1,2), has made its way as a widely used method in molecular and cell biology in both academics and industry. Pharmaceutical and biotech companies have set up libraries for large-scale screens employing thousands of short-interfering RNAs- (siRNAs) or short hairpin RNA- (shRNA) encoding vectors to identify new factors involved in the molecular pathways of diseases (3). The offered libraries can target either the whole genome of various organisms or just a subset of it. Based on these siRNA or shRNA libraries, RNAi screening enables massive parallel gene silencing to reveal the extent to which interference with the expression of specific genes alters the cell phenotype, and it is increasingly being used to reveal novel connections between genes and disease-relevant phenotypes (4–7).
Obviously the silencing efficiency of an RNAi reagent is very much dependent on its actual sequence (and structure) hence their design is of central importance and a field of intensive research. To date, a large number of target prediction computer programs and databases have been developed, such as Amarzguioui (8), Deqor (9), siR (10) E-RNAi (11), siRecords (12) and Rational siRNA (13) for RNAi construct design. In addition, several resources have been established to systematically collect and describe both experimentally validated RNAi reagents and related phenotypic results, such as GenomeRNAi (14) for human, Caenorhabditis elegans and drosophila; sIR (10) for a variety of organisms; FLIGHT (15) for drosophila; RNAiDB (16) for C. elegans and PubChem (17) for variety of organisms. Nevertheless, these resources rather help people who want to perform small/middle scale experiments and are of limited help for those who use large libraries from commercial providers (who often use proprietary, non-public algorithms for their construct design). Due to technical/practical reasons, there exists always a time-gap between the design and the actual production/delivery of the reagents. Furthermore, due to the relatively high costs of the reagents, researchers need to use a certain library typically for several years. Consequently, as genome annotations are still changing, there is a need to reanalyze the libraries with latest NCBI reference sequences (RefSeq) (18). For the correct interpretation of RNAi experiments, and also for measuring the quality of reagents, reagent-to-gene-model linkages/relations must be reanalyzed in regular intervals. Reagent designs with old RefSeq version may lack homology to any known mammalian gene in the latest RefSeq versions. Based on the new information, constructs may also match to different mRNA transcript sequence. Ultimately, RNAi reagent quality can serve to characterize unknown proteins and provide guideposts for follow-up analysis. RNAi databases with latest annotation and reagent knock-down specificity are essential for cross-correlating phenotypic screening information. At the same time, a comparison across multiple screens can also be used to evaluate and confirm the reliability of a particular RNAi reagent (side effects).
Companies typically offer three or more siRNA constructs per target and these can be used either as individual/single ones or can be mixed and used as pools. The main reason for this is the varying knock-down efficiency of the individual oligonucleotide and the occurrence of off-target or non-target effects. Since the discovery that not all siRNAs are equally potent in their ability to silence the gene products (18), a series of studies have pointed to a large number of ‘features’ that might be correlated to the higher efficiency of siRNA based silencing. Here we describe a publicly accessible database, the ‘RNAiAtlas’ with the task to track annotation evolution of siRNA reagents. RNAiAtlas has been designed for exploration and distribution of siRNA libraries updated with the latest genome annotation. Currently (beginning of 2012) the database contains 19 601 human genes including information about the three commercial siRNA libraries, such as sequence information, analyzed target specificity and predicted efficiency (10). In addition, RNAiAtlas is a pre-computed resource for the exploration and analysis of siRNA off-target associations. Optimal, user-friendly exploitation of siRNA off-target associations requires easy navigation between various displays so that not only the pairwise interactions, but also the network of interactions and the presence of potential (sub)modules in the network become visible. Previous RNAi databases listed above only present a single form of siRNA to transcript associations. Particular emphasis in RNAiAtlas has been placed on fast and easy navigation for off-targets, coupled to integrated visual outputs. The graphical representation of the network of siRNA to off-target gene connections provides a powerful tool for knock-down linkage analysis, facilitating the interpretation of siRNA screening results.
The database
Content
The database integrates the information of the siRNA reagents from three human, genome-wide libraries (beginning of 2012). It contains all together 196 024 siRNA constructs that have been designed by three major library suppliers (Applied Bioscience—Ambion, Qiagen, Thermo Scientific–Dharmacon). These whole-genome siRNA collections cannot be exhaustive and totally accurate unless gene databases are stable (18). The libraries have been analyzed and re-annotated with the RefSeq database, and the results for three RefSeq versions (2003, 2006 and 2011) are currently integrated. Annotation from different data stamps allows tracking of the siRNAs annotation evolution for target genes. Nevertheless, including ‘only’ the historic/newest annotation information is not sufficient to understand the siRNA specificity related issues, hence the results of a so-called ‘on-target analysis’ were also included in the database. These analysis results for the individual oligos are: the number of target transcripts, off-targets, target position on mRNA, pseudogenes, non-targets and wrong predicted siRNA (see the section below for a detailed description). A link to PubMed with a pre-formulated GeneID query search is also made available to allow the user to easily check for RefSeq RNA records relating to the gene of interest. An siRNA designed for a specific gene can have a variety of off-target connections as well. These connections are conceptualized in RNAiAtlas as networks and the size and organizational complexity of these networks present a unique opportunity to view a given library as something more than just a static collection of distinct siRNA constructs.
Principally, RNAiAtlas is designed to serve two main purposes (Figure 1): (i) it provides a genome-wide scale data set of siRNAs with consistent specificity ratings and (ii) it helps experimental RNAi researchers directly by visualizing off-target interactions between siRNAs and target genes. Thus, it can help bioinformatics scientists to interpret more reliable siRNA design tools.
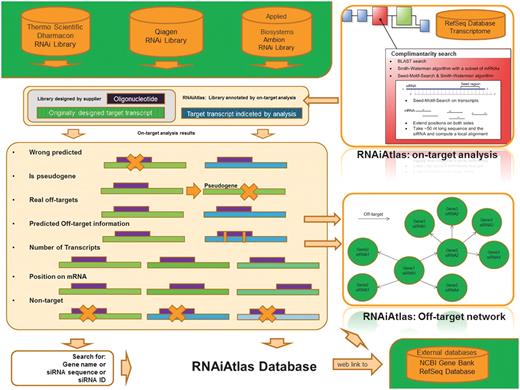
Overview of RNAiAtlas database content. The re-annotation of commercially available human genome-wide siRNA (three human, genome-wide) libraries with different NCBI reference sequences (RefSeq) were collected in a database. On-target analysis calculations were performed using a dedicated design/evaluation pipeline to quantitatively assess the specificity of the constructs. Interactive network visualization allows the inspection of the off-target network between an siRNA and its target genes.
On-target analysis
The specificity of an siRNA construct is a crucial factor in any silencing experiment (19). Protein expression silencing through the RNAi machinery works perfectly if the siRNA is totally complementary to its target mRNA. It is well known that single nucleotide mismatches between the siRNA and the target mRNA decrease the rate of mRNA degradation (20,21). The algorithms of the different companies for generating the best siRNA sequence typically take this into account and check and exclude siRNA sequences that have total complementarity to other than the target mRNA. Nevertheless, with evolution of the gene annotation and new releases of the RefSeq database (18), already designed siRNAs, today may match or not match to new mRNA transcript sequences. Hence, existing libraries need to be regularly analyzed for their specificity based on sequence-dependent analysis. We call this process ‘on-target analysis’ and the results are integrated into RNAiAtlas.
On-target analysis uses the latest reagent annotation and calculates the related specificity based on a complementarity search algorithm (Figure 1). All calculations were performed using a customized design/evaluation pipeline, HCDC-KNIME (22).
Details of the complementarity search
The aim of the analysis is to determine whether there exists a complementary region between the selected siRNA sequences and the mRNAs. Many different sequence alignment algorithms could be used for such complementarity search, but they are they are not necessarily optimal for this purpose by default. In our approach, we used/combined three different strategies to find nearly exact complementary regions as well as small local complementarities.
BLAST search
BLAST is a very efficient tool to find out if an obvious on-target siRNA exists with a full identical nucleotide sequence to the mRNA. Therefore, the BLAST search against the mRNA database from the RefSeq project is the first strategy for a complementarity search in our analysis concept. The BLAST analysis results for each siRNA oligo in a list of mRNAs were ranked according their sequence similarity score (length of similarity). The maximum value for this score is 19. Despite the full length similarity between the siRNA and mRNA, there can be still local mismatches between them. Though theoretically these could have been analyzed also by BLAST, but this would have required large computing capacity, so we rather used an alternative approach and performed an analysis based on the Smith–Waterman algorithm.
Smith–Waterman algorithm
The Smith–Waterman algorithm is an accurate algorithm to build local alignments between two sequences. Since its use with all mRNAs from the database is not practical, based on some empirical tests we decided to apply it only to the top 200 mRNAs from the BLAST results list (see Figure 1 RNAiAtlas: on-target analysis).
The result of this analysis is an alignment score with a maximum value of 95. In the case of a single nucleotide mismatch, the score would be approximately 86.
Seed-Motif-Search combined with the Smith–Waterman algorithm
Because of the mentioned runtime problem when performing a local alignment with the Smith–Waterman algorithm, a third variant to search for complementarity is introduced here. In this variant, an initial step reduces the length of the mRNA sequences to enable the use of a local alignment algorithm. At the beginning, all occurrences of the seed motif of every siRNA are localized in the genes. After detecting this small region, a sequence of ∼50 nt around this seed motif is cut out in the mRNA. Thus, as a result of this first step, a huge number of sequences of ∼50 nt in length are obtained containing the seed region of each siRNA. Due to the small length of the sequences it is now possible to perform a local alignment with the Smith–Waterman algorithm. The advantage of the Seed-Motif-Search for on-target analysis is that it limits the results to those genes which perfectly match with the seed region of the siRNA.
In the current version of RNAiAtlas, we concentrate only on siRNAs that had a minimum alignment score of 95 and length of full match of 19 nt (out of 21), meaning that we do not consider the 3' TT overhangs on position 20 and 21 (21). Such siRNAs without tolerating local mismatches are in principle ‘on targets’ but of course they may target not (only) their originally intended mRNA. In this case we were classifying them in the following four categories:
Wrong predicted: if oligonucleotide do not target gene annotated by supplier but target different gene.
Is pseudogene: if target gene is a pseudogene.
Real off-targets: if in addition to originally designed target gene, oligonucleotide also targets another gene (minimum alignment score 95 and length 19 to mRNA of gene annotated by supplier and in addition to mRNA of another gene).
Non-target: is true for siRNA if there is no target gene with sequence complementarity in entire genome for designed reagent (no mRNA transcripts where minimum alignment score is 95 and length 19).
In addition, for all oligos we calculated/provide the following information:
Number of transcripts: provides information about the number of transcripts of the targeted gene.
Position on mRNA: displays target site identification as a position of siRNA on mRNA sequence (5’, CDS or 3’ on mRNA).
This pre-computed results list of siRNAs (alignment score 95 and length of full match 19, based RefSeq 2011) enabled the selection of 26 393 siRNAs in case of Ambion library, 71 593 in case of Qiagen library and 71 764 siRNAs in case of Dharmacon library. On an average, ∼7% siRNAs per library are labeled as non-target reagents, 1% labeled as wrong predicted, 3% labeled as off-target and 1% targets being pseudogene. 55% target 1 mRNA transcripts, 17% 2 mRNA transcripts, 7% 3 mRNA transcripts, 21% more than 3 transcripts. Figure 2 demonstrates a list of three selected siRNAs for each library, their sequences and on-target analysis results.

Example of selected siRNAs and their on-target analysis results are shown from Qiagen, Ambion and Dharmacon libraries, analyzed with RefSeq 2012. The columns can be classified into five functional groups that contain information about: original target, transcripts information, off-targets, wrong predicted and on-target. The table illustrates all possible on-target analysis results that are incorporated into RNAiAtlas. The table data is generated off-line and fed into RNAiAtlas.
Off-target network analysis
For siRNAs that were classified as ‘real off-target’ in the previously presented analysis, we offer an additional visualization tool. For a given ‘real off-target’ oligo all of its targeted mRNAs are shown together with the other siRNAs that target these mRNAs. If one of these ‘other’ siRNAs is also classified as ‘real off target’, the network is extended with its corresponding connections.
Database user interface and implementation
User interface: Search
At the main page, the user can query a gene by entering the gene symbol, gene ID (GI), siRNA sequence, siRNA ID and selecting a year of RefSeq annotation (Figure 3a). The same search field also provides a batch query mechanism for a list of genes or siRNAs separated with a delimiter (comma, semicolon, space or tabulator). This allows users to copy–paste genes or siRNAs from text tables
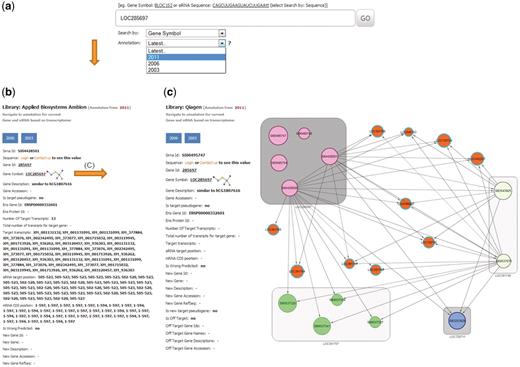
Example of a database search for gene LOC285697. (a) The search page allows for gene, siRNA sequence or siRNA ID. The search page provides a combo-box with selection possibility for the year of RefSeq database used for on-target analysis. Here LOC285697 as gene symbol was queried with the selected year of 2011 for RefSeq annotation. (b) Results card showing siRNAs, annotation for the queried gene and specificity parameters. (c) An example of the off-target network view in RNAiAtlas, centered on the query siRNA AGGCAGCAACAAGGATGGGAT (SI00495747) of gene ‘LOC285697’ from Qiagen human genome wide library (Network URL: http://rnaiatlas.ethz.ch/index/network/gene/LOC285697/library/3). Nodes in square (eg. gray squares) are individual siRNAs of same gene. Each node may have connection to off-target gene (represented again by three or four siRNAs). If off-targets genes are not existing in RNAiAtlas (e.g. pseudogenes, discontinued genes) siRNAs are having one edge to off-target gene (orange nodes).
User interface: Output
Upon initiating the search, the database retrieves all mapped siRNA probes and displays them as a card with navigation buttons for the different annotation years (Figure 3b). The record will present all relevant information about the siRNA, including the siRNA sequence, name of supplier, gene target information and target specificity parameters (Figure 3b). This can be used to find all siRNA constructs from the available libraries and various annotation years that overlap with a query. The pages of all siRNAs, from all suppliers targeting the same gene, and all their related parameters are displayed in the same view. The same card also displays the results of the calculated on-target analysis. For siRNAs showing an off-target effect, a fully interactive network display is available by clicking on gene name—allowing navigation through the combined off-target associations (Figure 3c). The network display allows iteration—zooming out of a particular module and visualizing its connections to other siRNAs or genes. From this network, pop-up windows lead to detailed information on each node (or edge) in the network, providing accessory information on an siRNA. The network display can be modified by adding or removing siRNAs, changing the required confidence level and by selecting or de-selecting certain evidence types.
As another feature of the user interface, permanent URLs can be retrieved for almost all pages served by RNAiAtlas—this facilitates cross-linking and archiving and also indexing by search engines and meta-sites. One central aim of the RNAiAtlas project is to achieve and maintain cross-connectivity and integration with other public resources in a user-friendly manner. Notably, such cross references do not have to be limited to simple text-based HTML links. Instead, partner websites can embed minimized icon-previews of RNAiAtlas networks.
Database implementation
We designed and implemented RNAiAtlas using Model View Controller (MVC) Zend Framework as back-end component. MySQL was used as the underlying relational database to store the library information and the related analysis results. At the front end, Apache Server 2.2.x is deployed to handle user requests and to forward them directly to controllers. To provide a user-friendly front-end, JavaScript and AJAX (Asynchronous Javascript and XML) as jQuery framework were adopted to program the client-side functionality and Cytoscape Web was used to create the interactive network components. All source code development was performed in Eclipse v3.6.0 (http://www.eclipse.org).
On-target analysis was performed using a custom design/evaluation pipeline, HCDC-KNIME (22).
Conclusion and future perspectives
To our knowledge, today this database is the largest collection of human siRNA reagents and their latest annotation, specificity information and off-targets connections. It was designed to assist experimentalists in determining which siRNA to use to inhibit their gene of interest or to check design quality for existing siRNAs. It is difficult to investigate information about siRNA constructs that failed or had poor knockdown without comprehensive bioinformatics analysis results integrated into database system. As more siRNAs are verified or new libraries will be available, this database will become increasingly useful for improving new siRNA design tools. Another future plan is to add and visualize secondary structure of siRNAs and their binding energy as a new parameter for specificity verification. The database design is prepared for import of other/additional RNAi (siRNA, shRNA, miRNA) libraries from other species. Currently for licensing reasons the siRNA reagents are identified by siRNA ID in the database. Sequences are password protected and available to users having access to the corresponding library license.
Funding
ETH Zurich Research Grants (ETH/0-20656-10 to UK), Switzerland.
Conflict of interest. None declared.
Acknowledgements
We are grateful to Andreas Kaufmann, Andreas Vonderheit for consultancy in RNAi field.

{kind=link}
{kind=link}
{kind=link}