





Abstract
The elucidation of the complex relationships linking genotypic and phenotypic variations to protein structure is a major challenge in the post-genomic era. We present MSV3d (Database of human MisSense Variants mapped to 3D protein structure), a new database that contains detailed annotation of missense variants of all human proteins (20 199 proteins). The multi-level characterization includes details of the physico-chemical changes induced by amino acid modification, as well as information related to the conservation of the mutated residue and its position relative to functional features in the available or predicted 3D model. Major releases of the database are automatically generated and updated regularly in line with the dbSNP (database of Single Nucleotide Polymorphism) and SwissVar releases, by exploiting the extensive Décrypthon computational grid resources. The database (http://decrypthon.igbmc.fr/msv3d) is easily accessible through a simple web interface coupled to a powerful query engine and a standard web service. The content is completely or partially downloadable in XML or flat file formats.
Database URL:http://decrypthon.igbmc.fr/msv3d
Introduction
Single nucleotide polymorphisms (SNPs) refer to a genetic change in which one nucleotide is replaced by another one and represent one of the most common forms of human genomic variation. Although SNPs are primarily associated with population diversity and individuality, they can also be linked to the emergence or the predisposition to disease, influencing its severity, its progression or its drug sensitivity. Several public repositories of SNPs exist, including GWAS Central (1), SwissVar (2) and dbSNP (3). Among these, dbSNP is probably the most extensive, with release 135 hosting more than 50 million human SNPs including 535 660 synonymous and 873 308 non-synonymous SNPs. The non-synonymous SNPs (nsSNPs), also called missense variants, are particularly important since they result in an alteration of the amino acid sequence of the encoded protein. Missense variants have been linked to a wide variety of diseases, for example by affecting protein function, by reducing protein solubility or by destabilizing protein structure (4). With the huge amount of protein information now available in various biological databases, including sequences, structures, functions, interactions, pathways, together with the development of in silico analysis tools, it is now possible to better understand the correlation between a missense mutation and the associated molecular phenotypes. Research groups have addressed this topic and have developed tools aimed at predicting the effects of missense variants on the function of a protein and its 3D structure, with varying degrees of success [for recent reviews, see refs (5–7)].
Over the last decade, numerous web servers have been developed to explore the effects of missense variants on characterized protein 3D structures and functions, including ModSNP (8), PolyPhen-2 (9), SNPs3D (10), StSNP (11), TopoSNP (12), LS-SNP (13), SNPeffect (14), MutDB (15), etc., which are publically available on the internet. These bioinformatics resources have different strengths and weaknesses (16). Although many of the web servers provide predictive analyses, according to a recent review (17), ‘most of the tested servers use NCBI’s dbSNP database as a primary source of SNP data, but are not up-to-date, increasing the chances that annotations for SNPs of interest will not be available to users’.
We have previously developed the SM2PH (from Structural Mutation to Pathology Phenotypes in Human) infrastructure (18), with the goal of contributing new useful bioinformatics resources dedicated to monogenic disorders for the research community. SM2PH-db was developed in the framework of the Décrypthon grid project (19), resulting from a collaboration between the AFM (French Muscular Dystrophy Association), IBM, and the CNRS (French National Research Centre) and involved the creation of a suite of an online analysis and visualization tools for the analysis of the correlation between genetic variations in disease-causing genes and the associated human phenotype. The initial version of SM2PH-db (18) contained about 2300 genes involved in human monogenetic diseases and has been regularly and automatically updated since September 2009. An integrative ‘genotype–phenotype relationship’ analysis was also performed, involving the characterization of how genetic alterations affect gene products (proteins) at the molecular level. Thus, the phenotypes associated with human pathologies were represented by their structural, functional and evolutionary context of all genes/proteins known to be involved in human diseases.
In this context, we designed SM2PH Central (Figure 1): a gene centric knowledgebase dedicated to the integration of and unified access to the information associated with any human protein (pathway, tissue expression, interactions, evolution, etc.). SM2PH Central provides access to a wide range of interconnected information that provides a global view from the gene to the phenotype. The system can be used to automatically generate any SM2PH database instance (i.e. a specialized database centred on a thematic use case, for example genes involved in a specific human disease, gene lists resulting from high-throughput analysis, etc.).
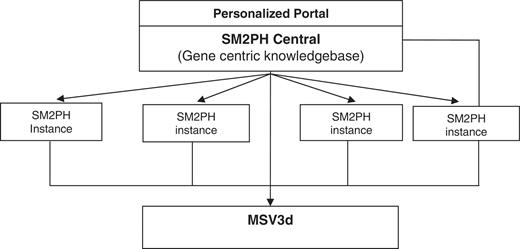
General architecture of SM2PH central. SM2PH Central allows the generation of SM2PH instances (focusing on specific sets of target genes) which can access variant information through the new MSV3d, devoted to human variant data and information.
Here we present MSV3d, a new database of previously identified missense variants involved in all human proteins mapped to 3D structure. MSV3d provides a unified access for SM2PH Central and its instances, for integration in any specialized database requiring interoperable and interconnected missense-related data and information, as well as for the biological community. The database is dedicated to automatic annotation of all human missense variants involved in 20 199 human proteins, thus covering all genes and diseases included in the Online Mendelian Inheritance in Man (OMIM) database (20). It facilities user exploration of the relationships between genetic variations and 3D structure via a unified access to databases, including SOAP web services, a Java API, simple queries and full or partial database download services. Statistical plots dynamically coupled with a powerful query engine allow the user to filter and analyse the data. In addition, the database also represents a useful benchmark set for any researcher who wants to develop and evaluate a machine learning method for classification or prediction of deleterious/neutral mutations. The database is automatically updated every 3 months and a major release is performed every year.
Description of database
Database content
The human missense variants in MSV3d are mainly retrieved from the dbSNP and SwissVar databases, but also from several locus-specific databases (LSDBs), e.g. the ALPL gene mutations database (http://www.sesep.uvsq.fr/database_hypo/Mutation.html). We classified all these variants in two main categories: disease-causing variant associated with OMIM diseases and Variant(s) of Uncertain Significance (VUS). In MSV3d, each missense variant is then characterized using four main levels of information:
Mutant information
This level involves data related to the gene and its associated protein, the chromosome position, the OMIM disease and genotype population reference. Pathogenicity prediction scores from external tools are provided by locally running the latest version of SIFT (21) and Polyphen-2 (9) to predict damaging effects of all missense variants in MSV3d. The SCOP fold classification (22) is also identified.
Conservation and physico-chemical changes
This level covers the information related to the mutated position in the context of its protein family. A multiple sequence alignment of the protein and up to 500 homologues [UniRef90 (23)] is constructed using PipeAlign (24) and annotated by MACSIMS (25). The MACSIMS annotation provides several descriptions of conservation, such as the conservation score of the substituted position, the percentage of mutated residues at the same position and the number of known mutations at this position. The physico-chemical changes induced by the amino acid substitution such as modifications in size, charge, polarity and hydrophobicity have been described previously (26). Modification of glycine or proline in the mutation is also identified. A global score reflecting the degree of modification induced by the substitution is also assigned. This score corresponds to the distance between the substituted residues based on a vector representation of the amino acids (see the MSV3d website for more details), where larger distances imply less conservative substitutions.
Structural features and modifications
These features include the structural annotations provided by MACSIMS, as well as detailed descriptions of the 3D context (e.g. residue relative accessibility, contact with an annotated site, etc.). Structural modifications induced by the amino acid substitution are predicted based on the mutant 3D models. These are automatically constructed using MODELLER (27) for missense variants that can be mapped onto a wild-type 3D model sharing >50% identity with the template used for the model construction. Secondary structures are deduced from the PDB (28) entry using the DSSP program (29). The effect in the protein relative stability upon single-site mutation is predicted with I-Mutant2.0 (30).
Spatial contacts
Four types of spatial contact have been defined: (i) the contacts between a residue and its direct 3D neighbours, based on the wild-type 3D model, (ii) the contacts between a mutant residue and its direct 3D neighbours based on the mutant 3D model, (iii) the contacts between residues in contact with the wild-type residue and their direct 3D neighbours, based on the wild-type 3D model and (iv) the contacts between residues in contact with the mutant residue and their direct 3D neighbours, based on the mutant 3D model.
Database statistics
MSV3d currently contains more than 445 574 missense variants mapped to 20 199 human proteins. Of these missense variants, 58 159 were found in SwissVar, 424 541 in dbSNP (build 135) and 37 209 in both SwissVar and dbSNP. A total of 24 379 the missense variants are considered as disease-causing variants and 421 195 as VUS.
Concerning the structural data, 10 713 structural templates from the PDB database have been identified allowing the mapping of 63 528 variants to a 3D structure. Among those mapped variants, 13 421 are identified in 265 SCOP fold classifications and 8023 variants are associated with 1479 OMIM diseases. Concerning gene conservation and function, 49 164 variants are mapped to one of the 2342 functional domains identified in the database (extracted from the Pfam protein family database (31), validated and propagated by MACSIMS) and 1799 HPO ontology terms from the HPO (Human Phenotype Ontology) database (32).
Up-to-date statistics concerning the physico-chemical changes induced by the amino acid substitutions, the conservation patterns, the localization in a secondary structure and/or functional domain are available on the ‘Statistics’ page of the website. Distributions of missense variants in SCOP folds or Pfam domains are also provided. As an example, Figure 2 illustrates the top 20 SCOP folds enriched in missense variants. By default, these statistics take into account the missense variants of all genes in the database. However, the user can also submit his own gene list in order to personalize the statistics analysis.
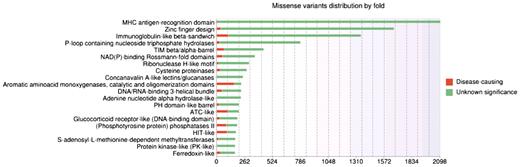
Histogram showing distributions of missense variants by SCOP fold. Each bar contains two parts: the red part represents deleterious substitutions and the green part represents tolerated substitutions.
Web interface and search engine
The MSV3d web interface (Figure 3) is designed to allow the user to rapidly query the complete database, for example by entering a protein name, gene name, SNP ID, OMIM ID, PDB template ID, chromosome position, protein fold or Pfam domain and to retrieve and export a list of missense variant data. MSV3d also provides a powerful full-text search service, allowing the user to search for any keyword stored in MSV3d without restriction to the index and field names. The results of a search can be visualized on the web or downloaded (Figure 3g) in a variety of formats such as XML or flat files. The user can also download the full database release in different formats.
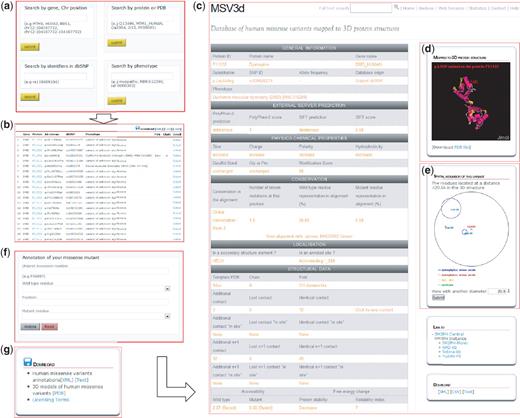
MSV3d web interface contains numerous functionalities including: (a) field search, (b) free text search, (c) detailed information, (d) 3D structure visualization using Jmol, (e) spatial neighbouring residue visualization, (f) missense annotation service and (g) download service.
To facilitate the structural analysis of missense mutation, we have incorporated the Jmol software (33) in the MSV3d interface. The Jmol applet is loaded automatically with an available structure model when a variant is selected on the web interface. Figure 3d shows the Jmol-based visualization for variations mapped onto the 3D structure. Mutations are automatically highlighted and neighbouring variants can also be identified.
Finally, the environment of neighbouring amino acid residues around a missense mutation is defined as follows: residues are considered to be neighbours of a mutation if they occur within a limited sphere in 3D space. Figure 3e shows the neighbouring residues of the p.Leu54Arg mutation in protein P11532 (with radius 20 Å).
Missense variant annotation with standard web service
The user can annotate a new missense variant using the web interface (Figure 3f) or using a programming interface via a SOAP web service. SOAP provides standard interoperability functions to communicate between applications running on different operating systems, with different programming languages. The SOAP WSDL protocol and API client of MSV3d (Java and C#) can be downloaded from our website (http://decrypthon.igbmc.fr/msv3d/cgi-bin/webservices).
Database construction
MSV3d pipeline
Taking advantage of the previous developments (18, 19), we have designed the MSV3d pipeline, involving more than 20 programs, firstly, to facilitate the investigation of the structural impacts of known or unknown missense mutations from all 20 199 human proteins, thus covering all known human genetic diseases and secondly, to guarantee a rapid update of the complete database content. The software pipeline has been deployed with high interoperability between all programs and their parallel application.
The schema in Figure 4 shows the main steps in the MSV3d pipeline. In general, the MSV3d pipeline takes a protein sequence as input and extracts associated missense variants from public databases. For each sequence, similarity searches are performed in public databases stored in the BIRD System (19), which is a local data warehouse supported by IBM DB2. BIRD provides a common architecture and relational schema for the integration of both local and public databases, as well as a unifying query system (BIRD-QL) for non-integrated data. The identification of background conservation and reconstruction of the evolutionary history of each reference sequence is based on Multiple Alignments of Complete Sequences (MACS) (34), thanks to a modified version of the PipeAlign program (24) and to the MACSIMS (MACS Information Management System) software. After PDB template selection and modelling where necessary, variant annotation is performed in the context of the 3D structure. The main steps in the process of MSV3d database creation are as follows:
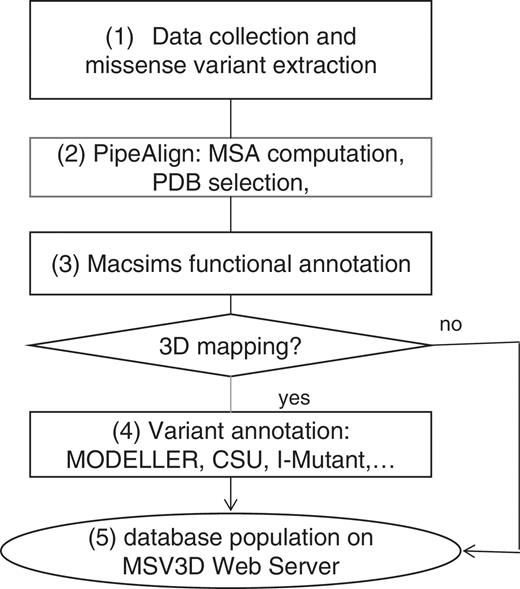
Schema of software pipeline.
Step 1: data input and missense variant extraction
This involved the implementation of automated protocols for the creation of a comprehensive collection of data related to (i) missense variants obtained from dbSNP, SwissVar and LSDBs, (ii) phenotypes obtained from the OMIM database. Most applications in the MSV3d pipeline use specialized BIRD-QL queries via the http protocol in order to automate retrieval, integration and mining of information in dbSNP and associated data such as proteins, genes and phenotype information.
Step 2: PipeAlign
The sequence analysis process (PDB selection, conservation, evolutionary information, etc.) has been automated using our in-house software cascade that has been shown to be robust and efficient (9). The PipeAlign cascade integrates eight programs to process: (i) protein sequence and structure database searches (Blast, Ballast), (ii) multiple sequence alignment creation [DbClustal (35)], correction [Rascal (36)] and quality estimation [NorMD (37), Leon (38)] and (iii) hierarchical classification into subfamilies [DPC (39), Secator (40)]. To address the challenges of the current sequence data deluge, a modified version of PipeAlign integrating a sequence sampling step (Sampler) after the Blast searches (41) has been implemented in the Décrypthon grid and recently in our local cluster at the Institute of Genetics and Molecular and Cellular Biology (IGBMC). More than 20 000 MSA are computed and indexed in the local data warehouse. The availability of a 3D structure or model of the protein is essential to gain insight into the structural impact of a missense variant. The best source of protein structural information is the PDB (28), which stores almost all the experimentally resolved crystallographic structures. 3D models of the wild-type proteins are automatically constructed by homology, using MODELLER (27). The models are built by inferring the structure of a protein (the target) from the structure of another putatively homologous protein solved by experimental methods (the template). Five homology models are constructed and the one with the best normalized DOPE score (27) is integrated in MSV3d.
Step 3: MACSIMS functional annotation
To characterize the background conservation and exploit different types of evolutionary data, we used MACSIMS to annotate the MACS with information such as: (i) taxonomic data, (ii) functional descriptions, (iii) known domains or domains similar to a known 3D structure, (iv) potential disordered regions, (v) blocks that do not correspond to disordered regions or known domains but that are conserved at the family or subfamily level and thus may constitute uncharacterized domains and (vi) conservation pattern of domains and residues. All the information associated with the MACS is collected and stored in XML format files.
Step 4: missense variant annotation
If the variant position is mapped to a 3D structure identified in Step 3, the structural context of each individual mutation is modelled based on 33 descriptors combining sequence/structure-related data using several software tools such as MODELLER, CSU (42), I-Mutant (30) (detail of the descriptors and computational software are provided on the MSV3d website).
Step 5
Finally, the full database is populated on the web server thanks to the BIRD-QL query engine, which is capable of managing the large volumes of heterogeneous data and provides up-to-date biological data for MSV3d.
Computer resource specification
To rapidly generate and update the very large database content, we use the Décrypthon grid and the IGBMC local cluster. The Décrypthon grid represents a total of 58 machines and 475 processors distributed on six nodes. The servers include multiprocessor machines (4–16 physical processors) under the AIX operating system and a cluster of single processor machines under the Linux system. To guarantee a permanent powerful CPU resource, we also deployed the complex software pipeline on a local cluster, representing a total of 16 machines and 240 processors. In order to facilitate the deployment of the MSV3d pipeline in the grid environment and local cluster, we developed interoperability protocols for the various programs and automatic procedures to compute, transfer and integrate the information from heterogeneous sources (software and biological databases) as well as to perform regular updates. Today, the complete update and annotation process for all human proteins (20 199 proteins and more than 400 000 missense variants) in MSV3d, takes up to 1 week.
Conclusions and future work
The large missense variant database mapped to 3D structures with regular updates is available for our scientific partners and for the wider community. Several access standards were developed to allow users to rapidly identify and retrieve the variant annotations. Facilitated access to such databases is an essential step to better understand how human genetic alterations affect the gene products at the structural level and subsequently to elucidate the relationships between genotypic and phenotypic variations. We have improved our original infrastructure and architecture in order to rapidly generate and manage the new data concerning all human proteins, thus facilitating an integrated approach to study any human genetic disease. The main advantages of MSV3d are (i) the full missense variant annotation for proteins without PDB structures, based on automated 3D modelling and (ii) the ergonomic and comprehensive database interface complemented with a SOAP-based remote API.
In the future, we plan to enhance the data integration by including structural surface topology descriptions using the M-ORBIS (for Mapping of mOleculaR Binding sItes and Surfaces) approach (43). This method, based on α-shape analysis, allows the precise mapping of different ‘functional’ regions such as the protein core and the non-interacting or interacting surfaces. The latter can then be further characterized as participating in homodimeric, heterodimeric, protein–peptide, protein–small peptide or protein–ligand interactions. With richer and more relevant knowledge, we hope to discover and extract pertinent relationships between missense variants and structural information using Inductive Logic Programming (44) or Support Vector Machine (45) approaches. We will also incorporate a novel formalism for the representation of protein evolutionary histories in the form of Evolutionary Barcodes (46). This new formalism allows the integration of different evolutionary parameters in a unifying format and facilitates the multilevel analysis and visualization of complex evolutionary histories. In the next major release of MSV3d, annotations for every possible amino acid replacement in the proteins will be integrated in the database. Finally, concerning data standards and semantic interoperability of biological data, we plan to implement a BioMart (47) interface to facilitate the exploitation and diffusion of our database in the biological community. More specifically, the standards proposed by the BioDBcore consortium (48) will be incorporated in MSV3d.
Funding
The work was performed within the framework of the Decrypthon program, co-funded by Association Française contre les Myopathies (AFM, 14390-15392), IBM and Centre National de la Recherche Scientifique (CNRS). We acknowledge financial support from the ANR (EvolHHuPro: BLAN07-1-198915) and Institute funds from the CNRS, INSERM, and the Université de Strasbourg. Funding for open access charge: ANR-10-BINF-03-02.
Conflict of interest. None declared.
Acknowledgements
The authors are grateful to Serge Uge for his assistance in implementing the local cluster. The IGBMC common services and platforms are acknowledged for assistance.

{kind=link}
{kind=link}
{kind=link}
{kind=link}